



IKZF1 N159Y 突变B细胞淋巴母细胞白血病/淋巴瘤:新亚型的临床和遗传特征
IKZF1基因改变包括部分或完全的基因缺失,在儿科和成人B-ALL中分别约占15%和40%,是预后不良的标志,然而IKZF1的体细胞点突变较为罕见。根据国际髓系肿瘤和急性白血病共识分类,IKZF1 N159Y突变(p.Asn159Tyr) B-ALL是一种新的罕见亚型,其发病率小于1%,此前仅有少数病例报告。
因此学者开展研究,首次全面提供了这种罕见亚型的临床、免疫表型和遗传特征,近日发表于《Haematologica》。


作者查阅了病理档案,与附近同事合作,并进行了全面的文献回顾,以识别携带IKZF1 N159Y突变的患者,总共确定了13例患者。研究分析了这些患者的遗传特征,包括常规细胞遗传学、荧光原位杂交(FISH)和全面的二代测序(NGS),后者包括RNA、DNA和拷贝数变异(CNV)的分析。通过NGS识别的变异分类依据美国医学遗传学与基因组学协会(ACMG)和分子病理学协会(AMP)的标准进行。所有在诱导治疗结束时收集的患者样本中定义的阳性微小残留病(MRD)通过流式细胞术分析确定为小于或等于0.01%,除了患者#10,其通过IgG重排的PCR分析进行。
患者人口统计学和临床特征
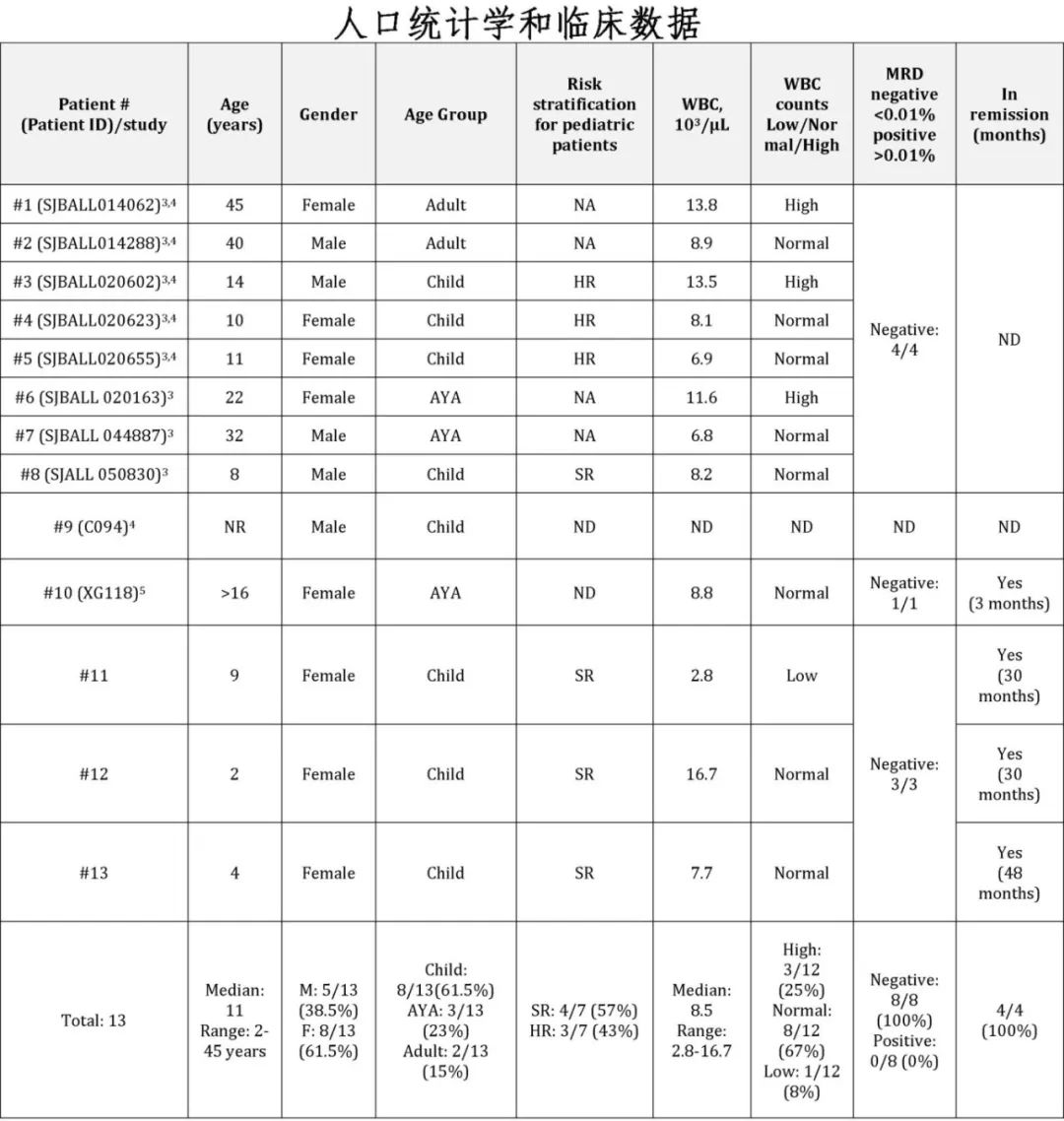
年龄和性别:患者年龄从2岁到45岁不等,没有明显的性别倾向,超过一半的病例发生在儿童患者中(61.5%,8/13)。
风险分层:儿童患者中包括标准危(SR)和高危(HR)患者(4例SR和3例HR)。
白细胞计数:大多数患者(67%,8/12)的白细胞计数在正常范围内。
临床表现:三例患者(#11-13)表现为贫血,但在诊断时没有白血病细胞的神经系统受累的证据。所有三例患者的原始细胞形态和免疫表型相似,表达CD10(强阳性)、CD19、CD22、CD34、CD38(较正常弱或阴性)、CD79a和TdT,而CD20、CD33和其他髓系及T细胞标记为阴性。
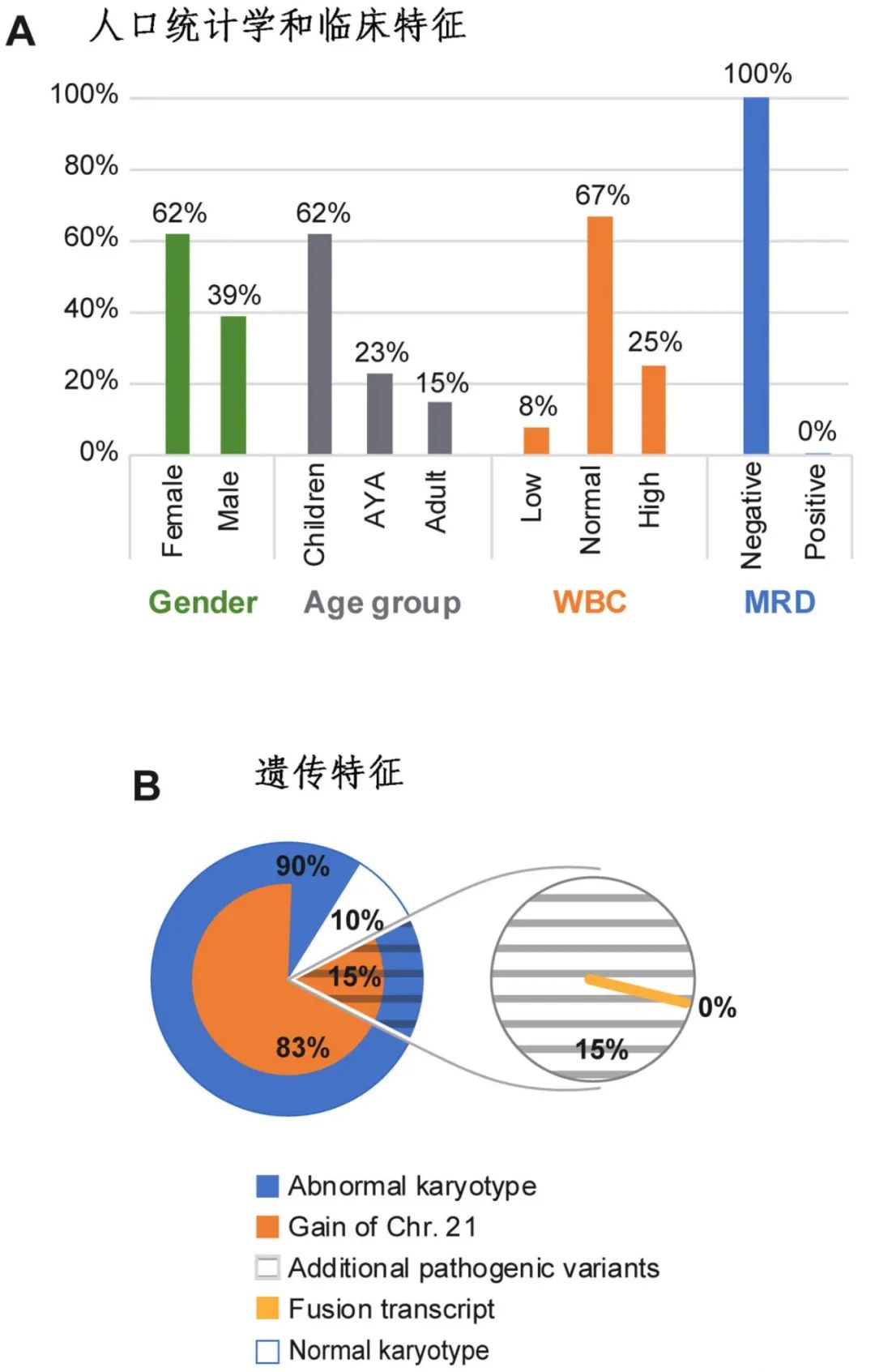
遗传特征
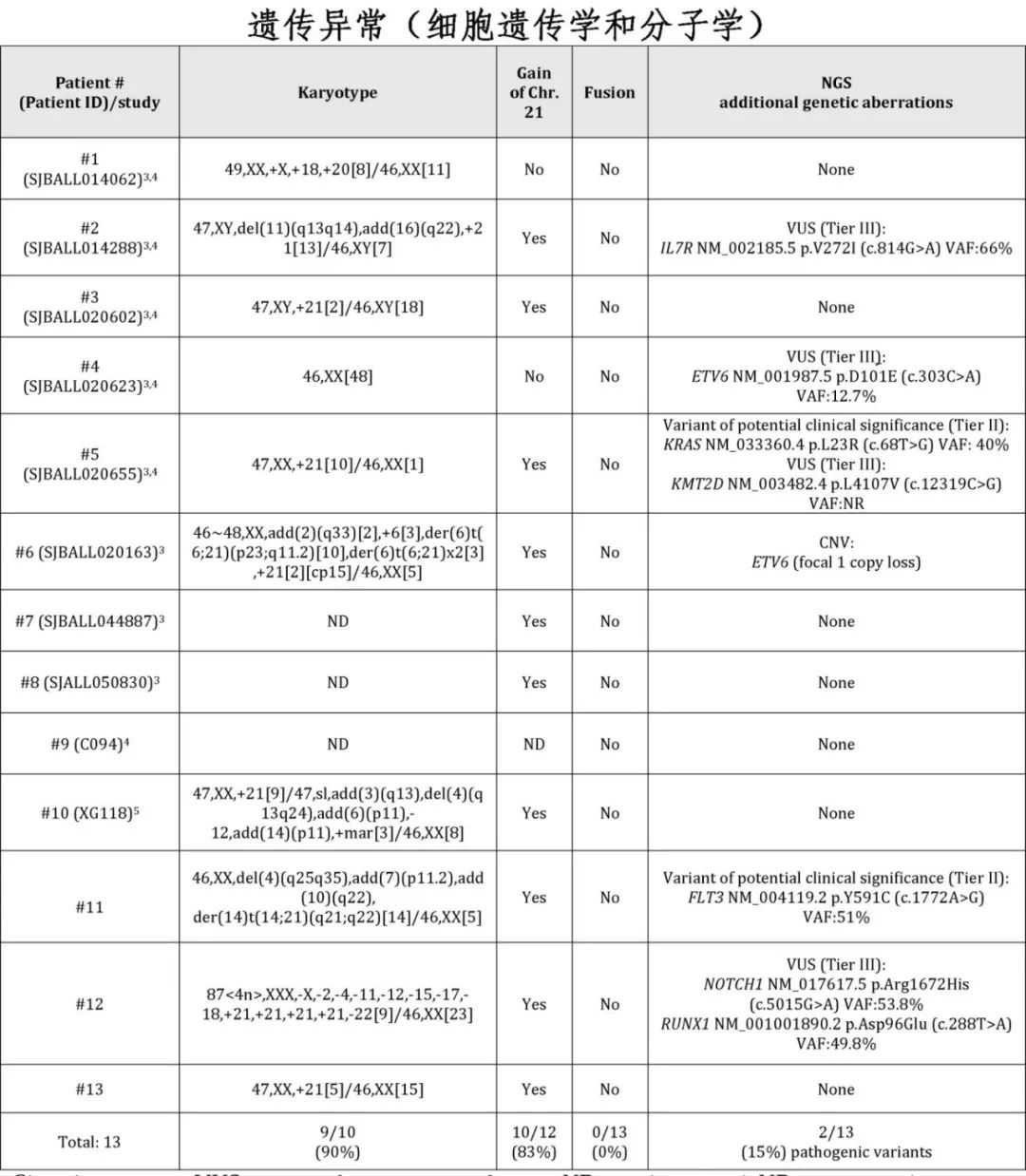
染色体分析:大多数患者(90%,9/10)存在异常核型,83%的患者(10/12)通过细胞遗传学分析和/或外显子测序的拷贝数变异确定为21号染色体增加。
附加突变:附加突变较为罕见,其中RAS信号通路相关基因(KRAS和FLT3)的致病变异最为常见(15%,2/13)。
融合转录本:未检测到融合转录本(0%,0/13)。
染色体易位:两例患者(#6和#11)通过染色体分析检测到染色体易位t(6;21)(p23;q11.2)和t(14;21)(q21;q22),但目前尚不清楚这些易位是否产生功能性融合产物。
治疗反应
所有在审查时已达到诱导治疗结束的患者均未检测到微小残留病(100%,8/8),并且达到完全缓解(100%,4/4)。
携带IKZF1 N159Y突变的B-ALL是一种罕见的复发性异常,更常见于儿童患者,并且与异常核型和21号染色体获得gain密切相关。此外,这些患者没有检测到融合转录本,附加遗传异常非常少,并且根据现有数据,所有达到诱导治疗结束的患者均未检测到微小残留病。
参考文献
Emily N. Alvey, Kai Lee Yap, Pamela Rathbun, Carrie Fitzpatrick, Shunyou Gong and Aida I. Richardson. B-cell lymphoblastic leukemia/lymphoma with mutated IKZF1 N159Y: clinical and genetic features of an emerging entity.Haematologica. 2025 Feb 27. doi: 10.3324/haematol.2024.286626
链接:http://www.lewenyixue.com/2025/03/14/IKZF1%20N159Y%20%E7%AA%81%E5%8F%98B%E7%BB%86%E8%83%9E%E6%B7%8B%E5%B7%B4%E6%AF%8D/

赶快来坐沙发