



肿瘤免疫中的“三折叠”:p53-IL-34-CD36信号轴介导的免疫逃逸
p53是人类癌症中最常见的突变基因。因为其在应激细胞中诱导细胞周期阻滞及诱导细胞凋亡过程中的作用,其又被称为基因组的“守护者”。p53的重新激活可以导致多种肿瘤退缩,这一过程需要免疫系统的参与。然而p53在肿瘤微环境中的调节作用目前依旧知之甚少。
今天小编和大家分享一篇近日由 中国科学技术大学魏海明教授实验团队 发表在 《Immunity》 上的一项研究。该研究揭示了在p53突变的肝癌模型中,肿瘤干细胞会过度分泌IL-34,通过IL-34-CD36信号轴调控巨噬细胞的脂肪酸代谢过程,使得肿瘤相关巨噬细胞向M2型巨噬细胞极化,引发T细胞免疫抑制,最终导致肿瘤免疫逃逸。研究中涉及的相关机制请见图1所示。
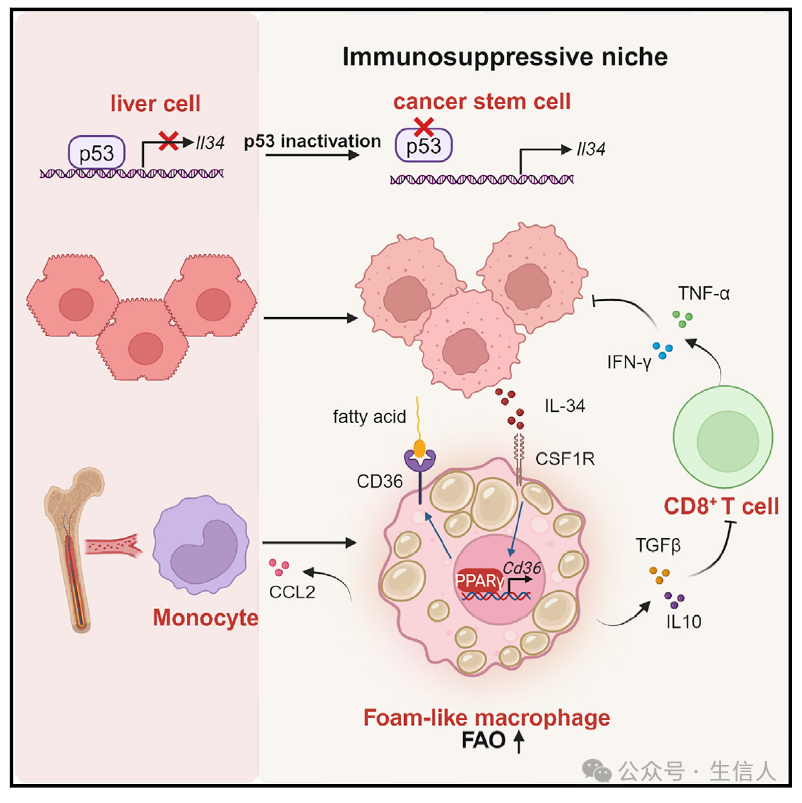 图1:p53失活引发的IL-34-CD36信号通路作用机制
图1:p53失活引发的IL-34-CD36信号通路作用机制
一、主要结果
1、“折叠第一面”--p53突变导致肿瘤干细胞分泌IL-34增多,促进肿瘤进展
为了理解p53在肿瘤微环境中的作用,作者使用CRISP-CAS9的基因编辑技术生成p53缺陷(Trp53null)以及野生型(Trp53wt)的小鼠肝癌模型,并对荷瘤小鼠的肿瘤进行RNA测序分析。作者注意到IL-34在Trp53null的小鼠模型中更高表达且与Trp53WT小鼠模型差异显著,这一现象也在qPCR和ELISA实验中所证实。
为进一步理解肿瘤微环境中,p53突变影响了什么细胞分泌IL-34增多?作者使用单细胞核测序技术(snRNA-seq)分别对Trp53null和Trp53wt的肿瘤进行检测,分析发现了肿瘤干细胞(CSCs)在p53突变后会上调IL-34的表达。多色免疫染色实验也证实IL-34和CSCs标志物EpCAM在p53突变的肝癌中大量聚集表达。
IL-34对于肿瘤微环境又有什么影响呢?作者在IL-34缺陷(IL-34-/-)的小鼠自发肝癌肿瘤模型中发现,IL-34的过表达会促进肝癌的进展。 这些结果表明IL-34在p53突变诱导的肿瘤进展过程中具有重要的促进作用(图2)。
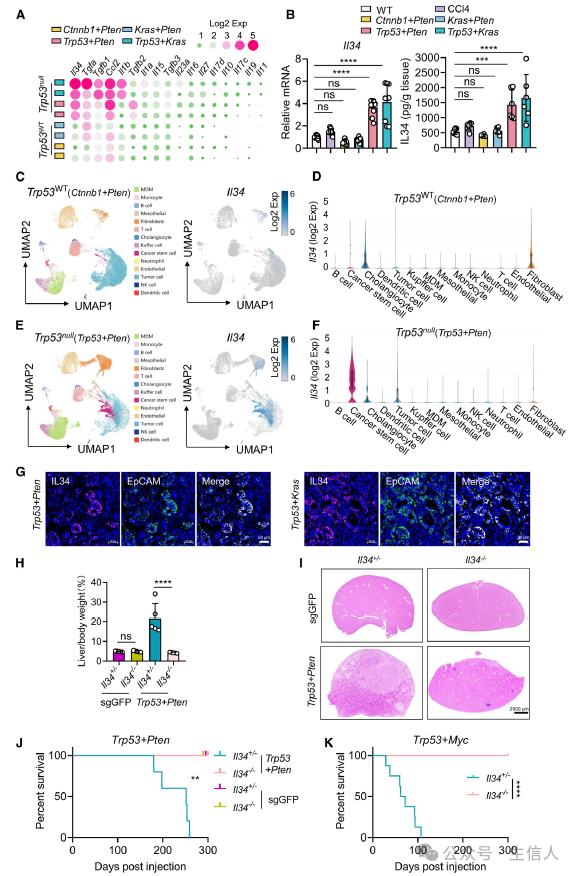 图2:p53突变使得CSCs分泌高水平IL-34促进肿瘤增长
图2:p53突变使得CSCs分泌高水平IL-34促进肿瘤增长
2、“折叠第二面”--IL-34通过CD36促进TAM细胞的脂肪酸摄取,使其向M2型巨噬细胞极化
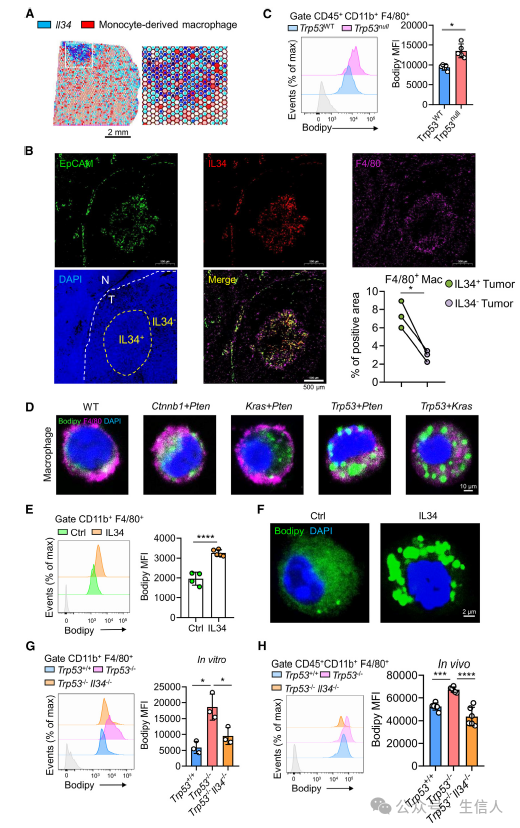
IL-34对于调控单核细胞增殖,生存具有重要意义[1]。作者通过单核细胞测序数据分析已然发现在p53突变的小鼠肝癌模型中IL-34和巨噬细胞同时表现出相较p53未突变的肿瘤模型更高的增长比例。之后通过10x空间转录组测序,作者发现单核来源巨噬细胞(MDMs)相关基因的表达分布围绕在IL-34阳性的细胞周围。多色免疫染色实验进一步证实了这一结论。
此外,研究者使用BODIPY脂肪酸探针对TAM中的脂肪酸进行检测,来自Trp53null肿瘤的肿瘤相关巨噬细胞(TAMs)比来自Trp53WT肿瘤的TAMs吸收了更多的脂肪酸,细胞内形成了更多的脂滴,TAMs泡沫化程度更高。此外,研究人员使用IL-34处理体外培养的骨髓源性巨噬细胞(BMDMs),发现IL-34会显著增加了细胞内脂肪酸的含量,并促进了脂滴的形成。 这些结果表明p53突变肿瘤模型中的TAM脂肪酸代谢重编程,形成类似于泡沫状巨噬细胞(图3)。
IL-34如何促进脂肪酸在TAM中累积呢?作者分别给予体外培养的BMDMs细胞IL-34过表达和正常处理两种处理方案,结果发现IL-34过表达的BMDMs细胞中会累积更多的脂肪酸。不同处理后的BMDMs的RNA-seq分析结果表明,CD36与IL-34的分泌密切相关。qPCR和流式细胞分析也证实了这一结论。CD36是与细胞中长链脂肪酸提取密切相关的蛋白。当使用CD36抑制剂(Sso)处理的时候,IL-34促进BMDMs细胞的脂肪酸提取这一过程受到抑制,表明IL-34对巨噬细胞的影响是通过CD36而发挥作用的。
有研究表明IL-4会使得巨噬细胞的脂肪酸氧化(FAO)代谢重编程,进而极化为M2型巨噬细胞[2, 3]。作者认为IL-34可能也能通过这一途径诱导巨噬细胞重编程。作者发现,过表达IL-34处理的BMDMs细胞会上调表达Mrc1,Arg1等M2型巨噬细胞标志分子;而当使用FAO抑制剂(Eto)或Sso处理后,巨噬细胞并不会朝向M2型巨噬细胞极化。 这些结果表明IL-34会通过CD36促进巨噬细胞的FAO重编程过程,并影响M2巨噬细胞的极化(图4)。
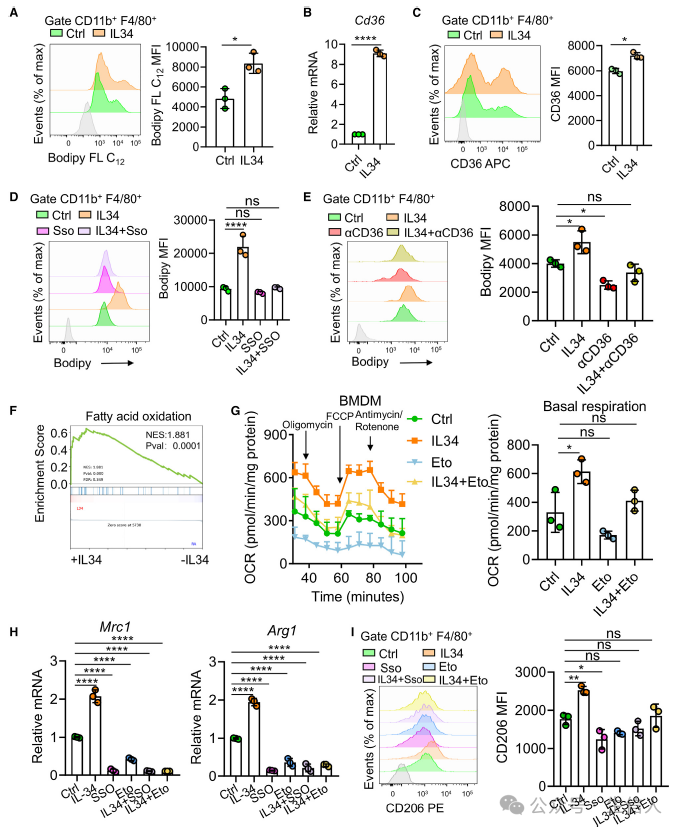 图4:IL-34 通过 CD36 促进脂肪酸吸收,并诱导巨噬细胞 向M2 型极化
图4:IL-34 通过 CD36 促进脂肪酸吸收,并诱导巨噬细胞 向M2 型极化
3、“折叠第三面”—p53突变肿瘤模型中M2型巨噬细胞介导的肿瘤免疫逃逸
IL-34影响TAMs的重编程这一过程,对于肿瘤进展过程中的免疫系统又有什么影响呢?作者发现在不控制小鼠脂质摄取的情况下,p53缺陷会加速肿瘤进展,而p53和IL-34同时缺陷时小鼠的肿瘤增长会受到抑制。当控制了脂肪酸摄取时候,p53介导的肿瘤进展过程不会因为IL-34的表达而发生影响。这些实验结果表明IL-34影响了巨噬细胞脂肪酸代谢重编程,进而促进肿瘤的免疫逃逸。
巨噬细胞会通过抑制T细胞功能介导肿瘤细胞的免疫逃逸。在p53缺陷的模型中,作者也发现了T细胞免疫抑制相关分子的上调表达,以及毒性分子IFN-γ和TNF-a下调表达。这些结果表明P53缺陷的肿瘤模型中T细胞处于免疫功能耗竭状态。
为了进一步明确TAM细胞在p53缺陷导致T细胞免疫耗竭中的作用,作者分选出脾脏T细胞与BMDM细胞以及p53缺陷或IL-34缺陷的肿瘤细胞共培养。结果表明,p53缺陷或者IL-34过表达均会使得T细胞比例降低,同时上调表达PD-1抑制性受体,下调表达IFN-γ和TNF-a活化受体。
在巨噬细胞中敲除CD36的条件性敲除小鼠肿瘤模型中,与巨噬细胞未敲除CD36的小鼠相比,肿瘤的增长受到抑制,且肿瘤浸润的T细胞比例显著增加。然而,当小鼠被注射CD8抗体时,其肿瘤的生长与对照组之间并未表现出明显差异。 这些结果表明IL-34-CD36信号轴在重塑TAM巨噬细胞极化过程中会抑制T细胞功能,进而介导肿瘤免疫逃逸(图5)。
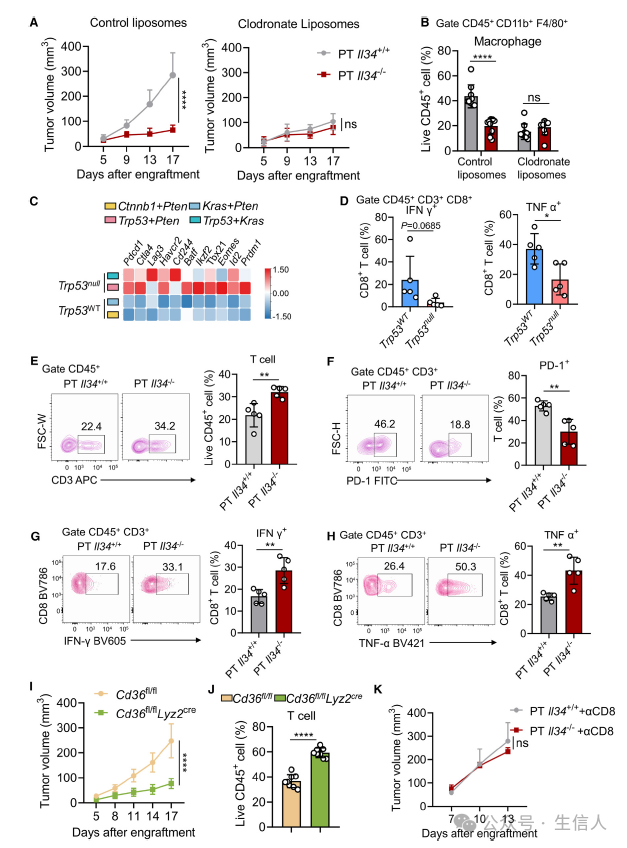 图5:IL-34 调控的 TAMs 抑制 T 细胞介导的抗肿瘤免疫,促进肿瘤免疫逃逸
图5:IL-34 调控的 TAMs 抑制 T 细胞介导的抗肿瘤免疫,促进肿瘤免疫逃逸
二、讨论与总结
p53作为一种重要的肿瘤抑制蛋白,在调控细胞周期,促进DNA修复和诱导细胞凋亡过程中具有重要作用。p53突变常见于多种癌症当中,但更多的研究关注于其在肿瘤细胞当中的影响,往往会忽视肿瘤细胞上的p53突变对于免疫细胞的直接影响。
在这项研究中,作者首次发现在p53突变的肝癌中,肿瘤干细胞会上调表达IL-34,驱动巨噬细胞脂肪酸代谢的CD36表达,引发巨噬细胞脂肪酸代谢重编程以及向M2型巨噬细胞极化,抑制T细胞免疫功能,最终导致肿瘤免疫逃逸。这一过程对于研究人员深入了解肝癌的免疫逃逸具有重要意义。然而,肿瘤干细胞在分化过程中可能会丧失IL-34的表达,这表明可能还有其他细胞参与这一通路。此外,关于肝癌的结论是否可以推广到其他癌症中,仍需进一步验证。
参考文献
[1] LUO W, ZENG Z, JIN Y, et al. Distinct immune microenvironment of lung adenocarcinoma in never-smokers from smokers [J]. Cell Rep Med, 2023, 4(6): 101078.
[2] VATS D, MUKUNDAN L, ODEGAARD J I, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation [J]. Cell Metab, 2006, 4(1): 13-24.
[3] HUANG S C, EVERTS B, IVANOVA Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages [J]. Nat Immunol, 2014, 15(9): 846-55.
链接:http://www.lewenyixue.com/2024/10/21/%E8%82%BF%E7%98%A4%E5%85%8D%E7%96%AB%E4%B8%AD%E7%9A%84%E2%80%9C%E4%B8%89%E6%8A%98%E5%8F%A0%E2%80%9D%EF%BC%9Ap53-IL-3/

赶快来坐沙发