什么!不吸烟的肺癌患者靶向治疗更不好
今天介绍的研究,考察转移性肺腺癌患者对靶向治疗的响应差异,该研究采取了全外显子测序,影像,以及小鼠模型体外培养后的单细胞DNA全基因组测序。研究发现:与仅携带 EGFR 突变的患者相比,携带携带 EGFR 和 TP53 两种突变患者的对 EGFR 酪氨酸激酶抑制剂(TKI)的治疗反应更可能存在异质性,这是由于全基因组加倍(全基因组加倍)和 TP53 共突变会导致基因组不稳定性和 EGFR TKI 耐药相关基因的基因组拷贝数异常增加造成的。该研究揭示了该如何彻底地研究肿瘤耐药性。

1)背景
肺癌是常见的癌症类型之一,也是癌症死亡的主要原因。大约85%的肺癌患者患有非小细胞肺癌(NSCLC),这是从未吸烟的患者中最常见类型。单独考虑,“从未吸烟”的肺癌是世界上第五大常见的癌症死因。在非小细胞肺癌中发现的最常见的基因突变是表皮生长因子受体基因(EGFR),它使癌细胞能够更快地生长。约10-15%的非小细胞肺癌病例中发现,特别是在从未吸烟的患者中。
针对这种突变的肺癌的靶向治疗,即所谓的EGFR抑制剂,已经有超过15年的时间。然而,虽然一些患者的患者的肿瘤在EGFR抑制剂的作用下缩小了,其他患者,特别是那些在p53基因(在肿瘤抑制中起作用)中也有额外突变的患者,没有反应并且生存率要差得多。但迄今为止,科学家和临床医生一直无法解释为什么会这样。
2)发现1:治疗后的混合响应普遍存在
研究者为了找到前述问题的答案,观测非小细胞肺癌患者靶向治疗开始时的基础影像扫描和治疗几个月后进行的首次随访扫描,这些扫描是针对只有EGFR突变或同时有EGFR和p53突变的患者。
图 1a 显示,在接受了厄洛替尼或化疗的肺癌患者中,约有 34% 和 24% 的患者出现了混合反应,即部分肿瘤缩小,同时其他肿瘤增大或出现新肿瘤。图 1b 展示了根据 Reiter 等人 (2019) 的定义,对 EORTC 数据库中接受厄洛替尼或化疗治疗的肺癌患者中混合反应的分析结果。在接受了厄洛替尼或化疗的肺癌患者中,约有 34% 和 24% 的患者出现了混合反应,即至少有一个肿瘤缩小 30% 以上,同时其他肿瘤增大或出现新肿瘤。这与图 1a 的结果一致,进一步证实了混合反应的普遍性。
图 1c 显示,与仅存在 EGFR 突变的肿瘤 (E) 相比,同时存在 EGFR 突变和 TP53 通路失活的肿瘤 (EP) 患者在接受奥希替尼治疗后,PFS 明显更短。这表明 TP53 通路失活会加速肿瘤进展。图 1d 和 1e 显示,在接受奥希替尼治疗的 EP 患者中,更可能出现混合反应,包括肿瘤增大或出现新肿瘤。这表明 TP53 通路失活会促进肿瘤对 EGFR TKI 治疗的耐药性发展。图 1f 显示,EP 患者在治疗过程中,不同肿瘤对治疗的反应差异更大,这可能是由于 TP53 通路失活导致的肿瘤异质性增加。
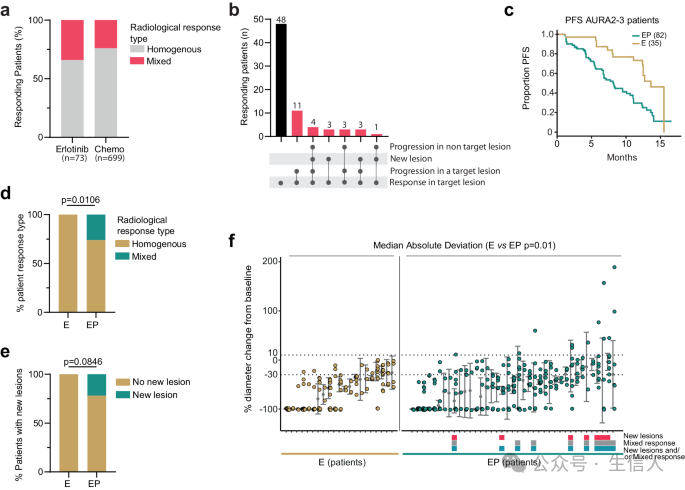
图1:治疗前后影像揭示混合反应普遍存在
上述临床数据提醒我们,TP53 通路失活会显著增加肺癌患者对 EGFR TKI 治疗的混合反应,并缩短患者的 PFS。这提示我们,在治疗 EGFR 阳性肺癌时,需要考虑 TP53 状态,并寻找新的治疗方法来应对 TP53 通路失活导致的耐药性发展。
3) 小鼠模型验证人类肿瘤中的药物效应混合反应
研究者之后使用基因敲除的小鼠模型,考察Trp53 通路失活与混合反应和治疗耐药性之间的关系。通过将小鼠中的基因对应到人的基因组上,图2a展示了小鼠和人类肺癌基因组中,与肿瘤发生相关的基因在染色体上的位置和拷贝数变异情况。粉红色代表染色体拷贝数增加,蓝色代表染色体拷贝数丢失。该图表明,在 E 和 EP 小鼠的肿瘤中,都与人类肺癌基因组中观察到的相同基因组事件相似,例如 AKT1 基因的拷贝数增加和 PBRM1 基因的拷贝数丢失。
图2b展示了 E 和 EP 小鼠在接受厄洛替尼治疗后,总生存期的差异。EP 小鼠的总生存期明显短于 E 小鼠,这表明 Trp53 通路失活会加速肿瘤进展。图2c展示了 E 和 EP 小鼠在接受厄洛替尼治疗一个月后,肿瘤大小的变化情况。EP 小鼠的肿瘤对治疗的反应差异更大,这表明 Trp53 通路失活会促进肿瘤的异质性。
图2d和e示了 E 和 EP 小鼠发生治疗耐药性的比例和耐药性发生的时间。EP 小鼠更容易发生治疗耐药性,并且耐药性发生的速度更快,这表明 Trp53 通路失活会降低肿瘤对治疗的敏感性,并加速耐药性的发展。
图2f该图展示了 E 和 EP 小鼠耐药性肿瘤中,发现的耐药机制。除了常见的 T790M 突变外,EP 小鼠的耐药性肿瘤中还发现了其他耐药机制,例如 EGFR 绕路突变和 Fgfr2 基因的获得性突变。这表明 Trp53 通路失活会促进肿瘤的克隆进化,并产生多种耐药机制。
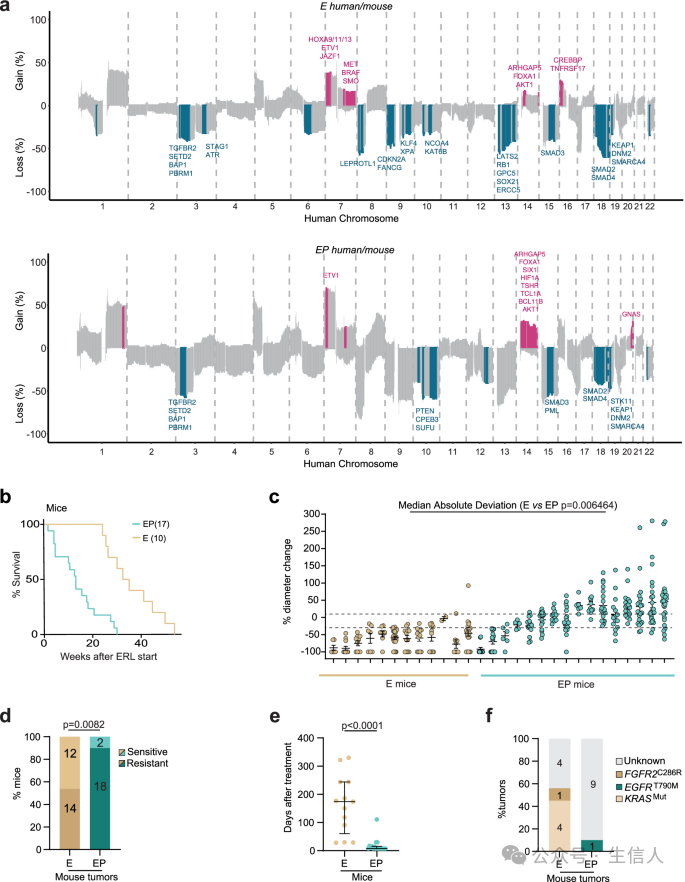
图2: 小鼠实验的结果展示
4)机制研究:单细胞全基因组揭示p53功能障碍与基因组不稳定性的联系
为了调查这些患者中一些肿瘤为何更容易产生药物抗性,研究团队随后研究了同时具有EGFR和p53突变的小鼠模型。他们发现,在这些小鼠的耐药肿瘤中,有更多的癌细胞加倍了它们的基因组,给予它们所有染色体的额外副本。 通过对小鼠模型中的癌细胞进行单细胞高深度全基因组测序,可进一步探查临床数据中的混合反应的成因机制。结果发现 Trp53 突变小鼠肿瘤中 SCNAs 的频率更高,且与 TKI 耐药性相关基因的拷贝数变化更频繁。
图3a显示了从E肿瘤和EP肿瘤中分离的单个细胞中,有多少比例的经过了早期基因组丢失。癌症细胞分数较低意味着细胞经历了早期丢失,并且在肿瘤中占较小比例。可发现EP肿瘤(具有p53功能障碍和全基因组加倍全基因组加倍)表现出比E肿瘤(仅具有p53功能障碍)更高的基因组不稳定性。这意味着EP肿瘤更容易发生染色体数量变化,如染色体丢失或增益。
图3b:显示了治疗前后E肿瘤和EP肿瘤中染色体数量变化(如丢失和增益)的频率。EP肿瘤中染色体数量变化的发生率更高,尤其是在MET、BRAF和Kras等与TKI耐药性相关的基因中。
图3c: 显示了EP肿瘤中与TKI耐药性相关的基因的染色体数量变化(如丢失和增益)的频率。EP肿瘤中这些基因的染色体数量变化的发生率更高,这可能导致耐药性。这表明全基因组加倍和p53功能障碍为耐药性发展提供了多种途径。
图3d显示了来自同一肿瘤和同一细胞质组的单个细胞的基因组异质性。EP肿瘤中的细胞间异质性更高,这表明全基因组加倍和p53功能障碍导致细胞之间发生更多不同的染色体数量变化。图3e显示了来自E肿瘤和EP肿瘤的每个肿瘤的香农多样性指数,这是一个衡量细胞间异质性的指标。EP肿瘤的香农多样性指数更高,这表明细胞间异质性更高。
图3f显示了来自E肿瘤和EP肿瘤的每个肿瘤的全基因组加倍频率。EP肿瘤的全基因组加倍频率更高,这表明全基因组加倍在EP肿瘤中更常见。图3g显示了来自E肿瘤和EP肿瘤的全基因组加倍频率。EP肿瘤的全基因组加倍的发生频率更高,这表明基因组不稳定性更高。图3h和i显示了两个外部数据集上来自E肿瘤和EP肿瘤的每个肿瘤的wGII,这是一个衡量基因组不稳定性的指标,结果EP肿瘤的wGII更高,这复现了之前的结果。
以上小鼠实验中的发现,针对只基于人源样本无法深入研究机制的缺陷,发现p53功能障碍和全基因组加倍导致细胞间异质性增加,这为耐药性发展提供了多种途径。这些发现有助于我们理解在EGFR突变驱动的肺癌中对靶向治疗的耐药性机制,并为开发新的治疗方法提供了见解。
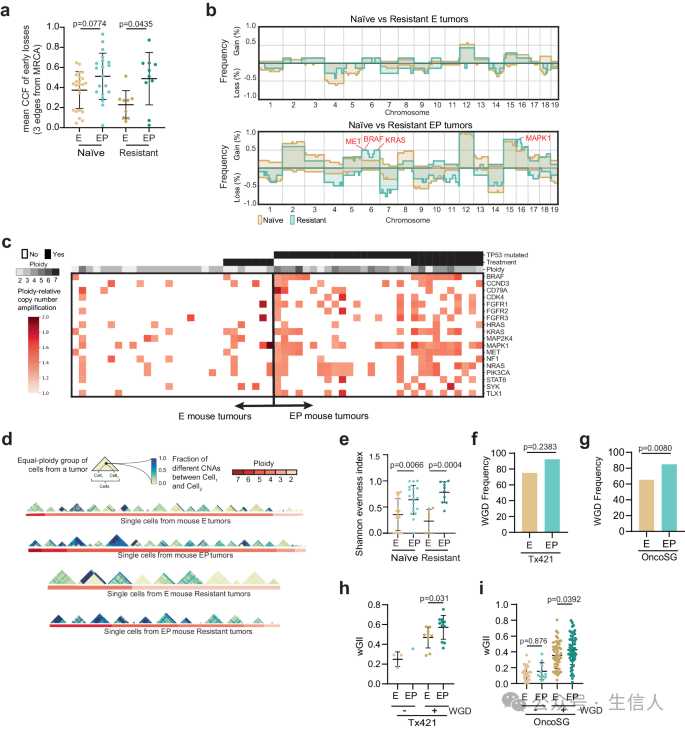
然后,研究人员在实验室中对肺癌细胞进行了EGFR抑制剂处理,一些只有单一的EGFR突变,一些同时具有两种突变。他们发现,在接触药物的五周内,具有双突变和双倍基因组的细胞显著更高比例地繁殖成新的耐药细胞。 之后研究者使用具有克隆 EGFR/TP53 突变的 PC9 细胞系,在存在或不存在 全基因组加倍 的情况下评估对 Erlotinib 的耐药性。结果发现在 PC9 细胞系中,全基因组加倍与 p53 功能障碍共同存在会促进耐药性克隆的出现,并且导致 SCNAs 频率增加和细胞间多样性增加。
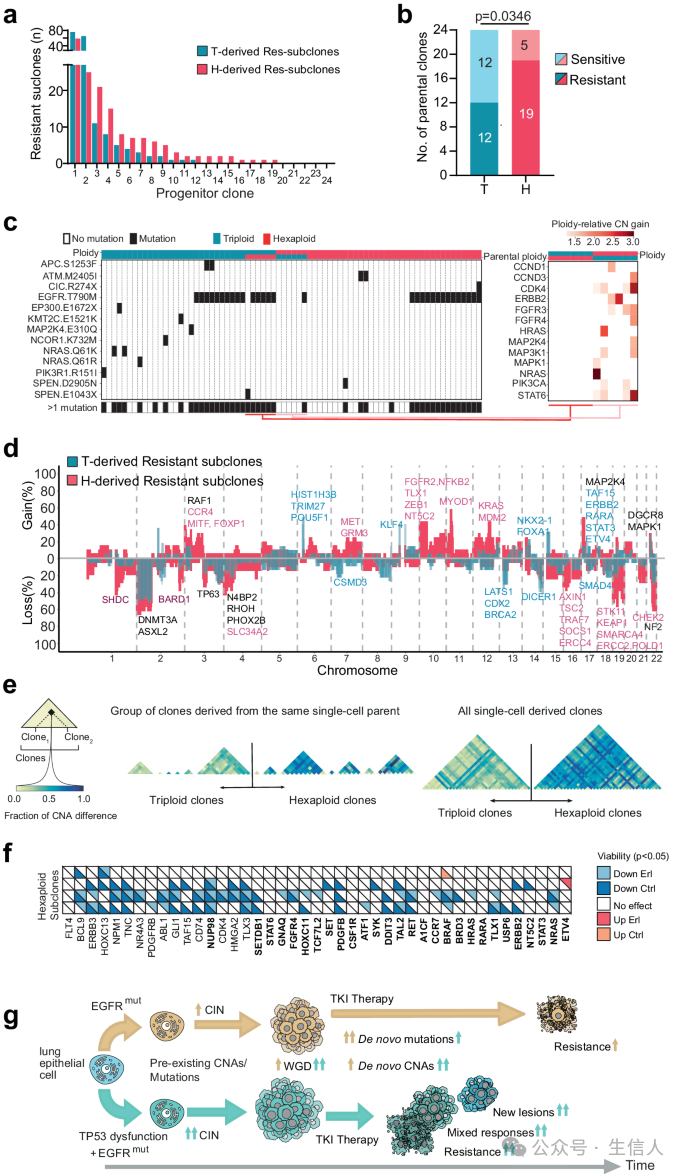
图4a展示了24个三倍体(T)和24个六倍体(H)PC9细胞系在5周厄洛替尼治疗后产生耐药亚克隆的数量。H细胞系产生耐药亚克隆的比例显著高于T细胞系,表明全基因组加倍和TP53功能障碍共同促进了耐药性的发展。
图4b比较了T和H细胞系产生至少一个耐药亚克隆的克隆数量。H细胞系中产生耐药亚克隆的克隆数量显著多于T细胞系,进一步证实了全基因组加倍和TP53功能障碍的协同作用。图4C图左侧展示了来自T和H细胞系的耐药亚克隆中,与EGFR通路相关的基因发生体细胞突变的频率。右侧展示了耐药亚克隆中,与EGFR耐药相关的基因发生拷贝数变化的频率。与T细胞系相比,H细胞系中耐药亚克隆中发生拷贝数变化的频率显著更高,表明全基因组加倍和TP53功能障碍导致SCNA更频繁地发生。
图4D图展示了T和H细胞系中,耐药亚克隆中发生拷贝数变化的频率。与T细胞系相比,H细胞系中发生拷贝数变化的频率显著更高,特别是在与EGFR耐药相关的基因中,表明全基因组加倍和TP53功能障碍导致SCNA更频繁地发生。图4E图展示了来自T和H细胞系的耐药亚克隆中,克隆间多样性(即具有不同体细胞拷贝数变异的克隆比例)的比较。H细胞系中克隆间多样性显著高于T细胞系,表明全基因组加倍和TP53功能障碍导致细胞间异质性增加。
图4F图展示了通过siRNA敲低耐药H细胞系中过度表达的基因后,对厄洛替尼敏感性的影响。10个基因的敲低导致至少一个H细胞系对厄洛替尼敏感,表明这些基因可能参与耐药性。图4G图展示了EGFR突变肺癌中,E和EP肿瘤中耐药性发展的模型。E肿瘤具有功能性的TP53,但发生全基因组加倍,主要通过点突变发展耐药性。EP肿瘤发生全基因组加倍且TP53失活,表现出更高的细胞间异质性,主要通过SCNA发展耐药性。耐药机制包括SCNA相关的基因拷贝数变化和点突变。
5)小结
该研究展示了为什么具有p53突变与非吸烟相关肺癌患者的更差生存率有关,这是由于EGFR和p53突变的组合使基因组加倍。这增加了通过染色体不稳定发展出耐药细胞的风险。非小细胞肺癌患者已经进行了EGFR和p53突变的检测,但目前还没有标准的测试来检测整个基因组加倍的存在。研究人员已经在寻求开发用于临床的诊断测试。
从方法学上来看:该研究采取了细胞水平的单细胞全基因组测序 (scWGS),基于WES的体细胞拷贝数变异分析,同源基因组加倍。结合了临床数据,小鼠模型和细胞系分析,一步步深入,推进到具体的分子机制。该研究的方法学可以为其他癌症研究和临床应用提供重要的借鉴,帮助我们更好地理解肿瘤异质性和耐药机制,并开发新的治疗方法。
从临床意义上来说:一旦我们能够识别出既有EGFR又有p53突变,并且肿瘤显示出整个基因组加倍的患者,我们就可以更有选择性地治疗这些患者。这可能意味着更密集的后续跟踪,早期放疗或消融以针对耐药肿瘤,或者早期使用EGFR抑制剂的组合,如奥希替尼,与其他药物包括化疗。
链接:http://www.lewenyixue.com/2024/10/18/%E4%BB%80%E4%B9%88%EF%BC%81%E4%B8%8D%E5%90%B8%E7%83%9F%E7%9A%84%E8%82%BA%E7%99%8C%E6%82%A3%E8%80%85%E9%9D%B6%E5%90%91%E6%B2%BB%E7%96%97%E6%9B%B4%E4%B8%8D%E5%A5%BD/