Nat Methods | 基于测序的11种空间转录组学方法的系统比较
导读
单细胞转录组测序(scRNA-seq)在剖析细胞异质性方面发挥了重要作用,目前已被作为研究关键生物医学难题的重要手段。但scRNA-seq在获取组织结构、细胞相互作用和功能状态所必需的空间背景方面仍存在不足。
为了解决这一挑战,空间转录组技术(Spatial transcriptomics, sST)应运而生,现在已经成为一种关键的补充方法,其可以在保留空间信息的同时实现全面的转录组分析。虽然基于sST技术已经取得了较大的进展,但目前还缺乏对不同平台进行全面的基准测试评估。
近日,广州国家实验室 田鲁亦团队 、西湖大学 刘晓东团队 合作在 Nature Methods 发表了题为“Systematic comparison of sequencing-based spatial transcriptomic methods”的文章。 研究团队选择了一系列具有明确定义的组织结构特征的组织作为参考,使用其生成数据比较了11种sST方法。 该研究有助于研究人员选择适合自身研究的sST平台,并有助于就评估标准达成共识,为未来的基准测试工作建立框架,进而可作为空间转录组分析计算工具开发和基准测试的金标准。
文章发表在 Nature Methods
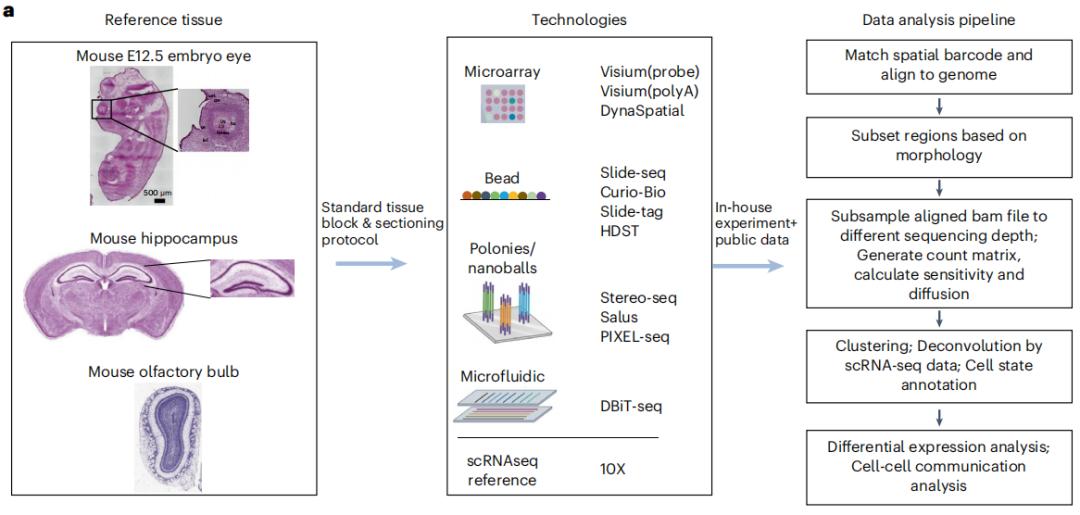
首先,研究团队基于不同的空间索引策略,对空间转录组学方法进行了系统的基准测试,包括 微阵列(基于探针和基于polyA的10X Genomics Visium、DynaSpatial)、基于磁珠的方法(HDST、BMKMANU S1000、Slide-seq V2、Curio Seeker、Slide-tag)、基于聚合酶链反应菌落或纳米球的技术(Stereo-seq、PIXEL-seq、Salus)和微流控技术(DBiT-seq) 。
此外,研究还选取了形态特征相对明确的成年小鼠大脑、E12.5小鼠胚胎以及成年小鼠嗅球作为参照组织。例如,E12.5小鼠胚胎中的眼睛具备已知的结构,晶状体被神经元视网膜细胞环绕,小鼠嗅球具有清晰的层状分离以及各类神经元类型,为进行 sST基准研究提供了理想的参考样本。研究团队在源自以上三种组织类型的35个实验中系统地评估了11种sST方法,并构建了一个标准的基准测试流程,以促使sST方法能够进行同质数据处理,展开公平比较。
针对评估空间转录组学方法面临的挑战,研究人员精心设计了系列基准研究来应对。首先, 研究选择了一组理想的参考组织样本,这些参考组织样本遵循以下标准 :该组织来自广泛使用的模式生物,并且大多数研究机构都能获取;该组织具有稳定的细胞类型和特异性标记基因表达;参考区域还应具有清晰的形态,并且在切片中易于被发现。连同参考组织,研究团队还开发了一种切片方案,能够帮助研究者在未来重现并生成可比较的数据。除此之外,研究团队还使用了多个基准指标和工作流程,使得在相同的组织区域比较不同的方法成为可能。最终, 研究团队在genographix.com 上生成了cadasSTre ,这是一个用于sST基准测试的跨平台数据集。研究团队在35个实验中对11种sST方法进行了系统评估。
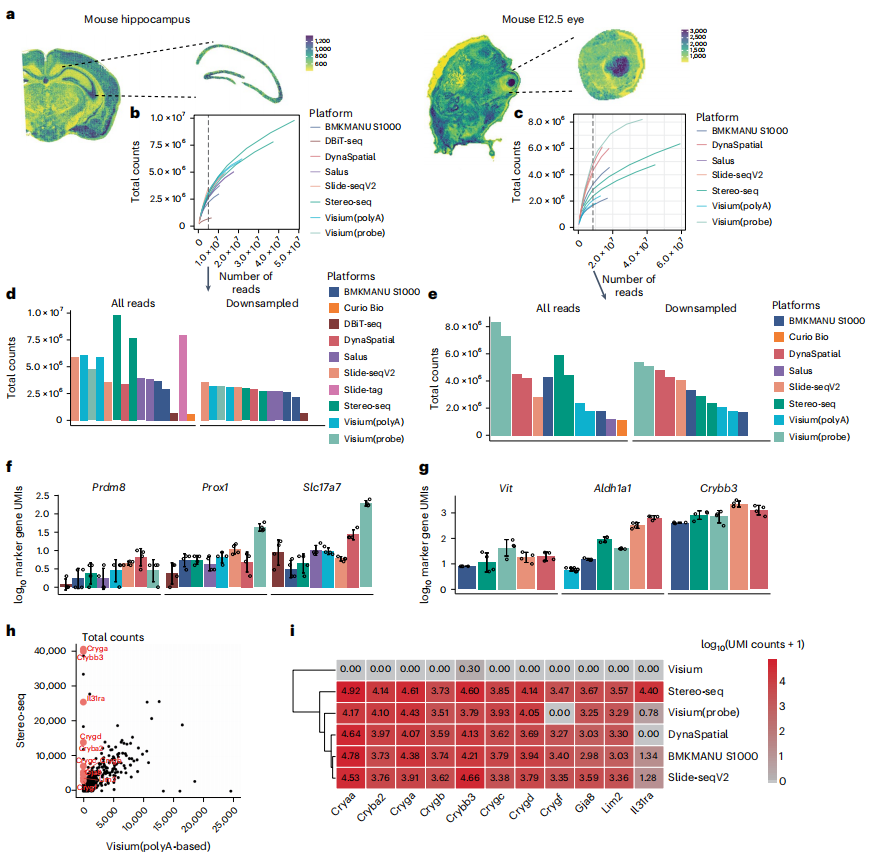
整体来看,该研究全面比较了各方法从基本指标到下游分析的各个方面的数据,包括灵敏度、聚类性和标记基因检测等,并总结了每个评估步骤中使用的方法。分析结果表明,空间转录组学需要更多的测序数据才能达到饱和,而研究中所使用的数据远低于饱和水平。 Stereo-seq、Slide-tag、Visium(基于探针)在原始测序深度下显示出更好的捕获效率, Slide-seq V2、Visium(基于探针)、DynaSpatial 在标准化测序深度下具有更好的捕获效率。
此外,研究人员 在基于polyA的Visium平台上发现了意外的基因捕获偏差 ,即其他技术始终捕获的标记基因在基于polyA的Visium数据中未出现。考虑到Visium是使用最广泛的商业平台,因此,在未来的评估比较研究中纳入其他参考组织中进一步验证其基因捕获偏差非常重要。
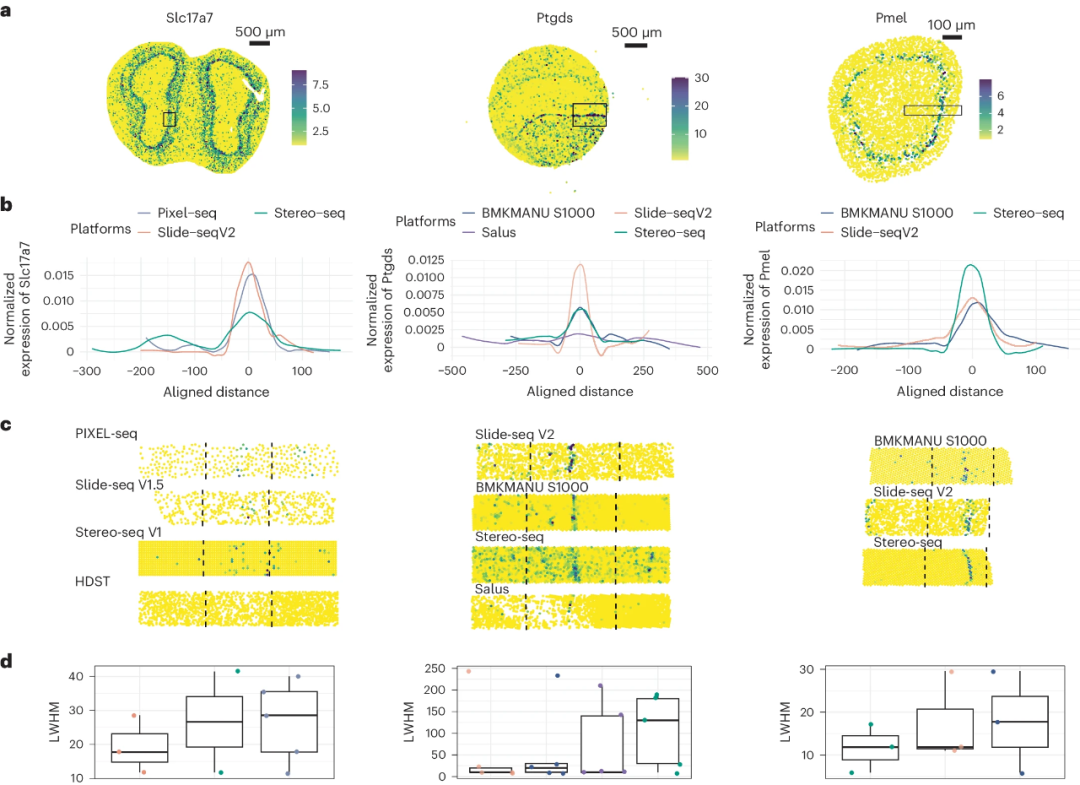
虽然spot大小已成为评估每种方法分辨率的一个重要指标。但该研究发现扩散(Diffusion)是影响实际分辨率的关键因素。此外,透化优化实验表明,改变透化时间对扩散有很大影响。具体而言, 在针对每种组织类型优化透化时间时,不同的技术在不同组织类型中表现出不同的扩散特征。 虽然某些技术能识别亚细胞spot大小,但由于灵敏度有限和扩散较高,其实际分辨率无法达到与具有真正单细胞分辨率的Slide-tag相同的水平。因此, 未来sST的进一步发展将受益于增强的扩散控制和改良的透化条件和时间。
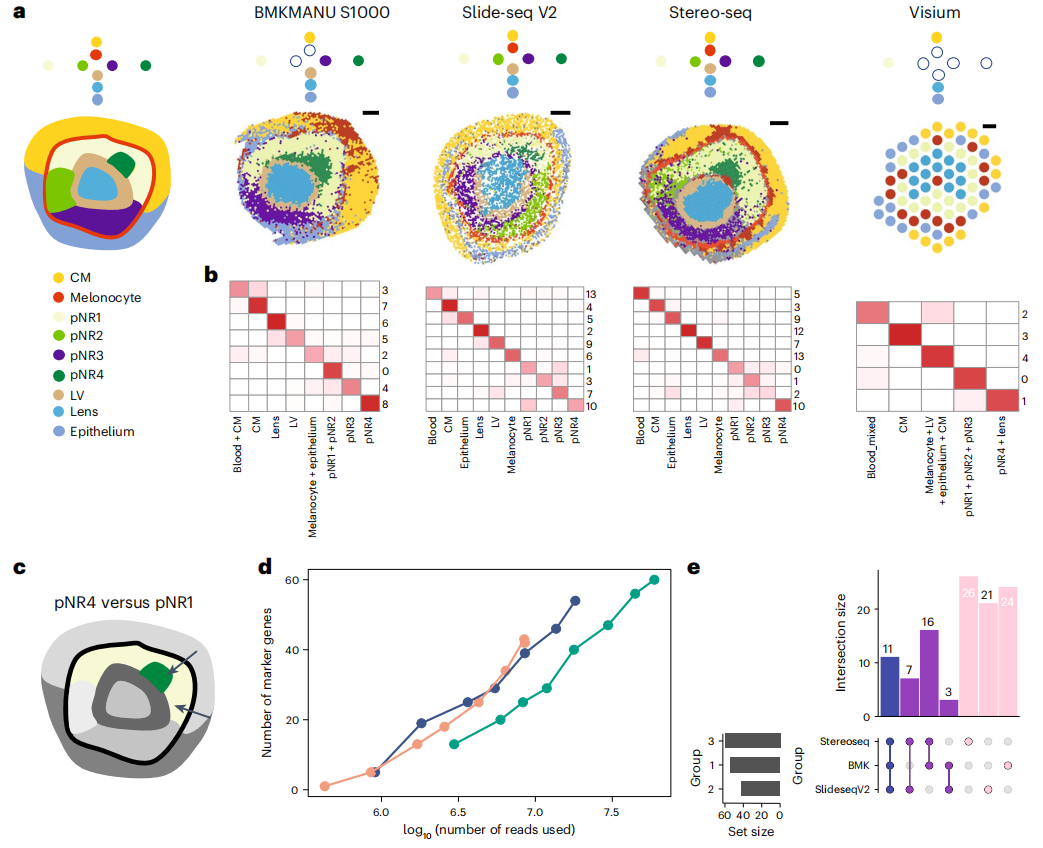
分析结果还表明, 为空间数据设计的聚类工具可能并不比单细胞的聚类方法表现更好 。换言之,虽然在某些数据集中纳入额外的空间和组织学信息会提高聚类的准确性,但与仅使用基因表达数据相比,这并不能始终如一地改善聚类结果。研究还发现, 来自单细胞维度的细胞注释可能无法产生详细的细胞状态,来自空间数据的聚类则可以给出互补的结果,并且其在解析具有空间模式的稀有细胞状态方面表现更好 。因此,在注释空间转录组学数据时,考虑有和没有单细胞转录组数据作为参考对于后续的分析很重要。
图5. 下游分析性能的比较
综上所述,该研究生成了sST方法的第一个系统基准方案。虽然研究团队努力展示所有技术的最佳性能,但仍有一些实验并未完全被优化,例如DBiT-seq和Curio Seeker,这意味着它们的数据可能无法代表其最佳性能。需要注意的是,sST实验通常很考验实验技能,因此真正上机测序前的操作效果不可忽视。
当前,sST领域正在迅速发展,每种技术的性能可能会随着时间的推移而进一步优化和完善。这也就要求领域内的研究人员持续评估不同工具的性能,以满足快速发展领域的需求。
研究团队表示,他们将继续维护和更新genographix.com上cadasSTre的在线基准数据库,并持续提供新的数据集。通过这个平台,来自世界各地的研究人员都可以访问最新的基准数据,贡献他们的发现,并与同行合作以推动该领域的发展。
You, Y., Fu, Y., Li, L. et al. Systematic comparison of sequencing-based spatial transcriptomic methods. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02325-3.
版权声明:本文为“乐问号”作者或机构在乐问医学上传并发布,仅代表该作者或机构观点,不代表乐问医学的观点或立场,不能作为个体诊疗依据,如有不适,请结合自身情况寻求医生的针对性治疗。
链接:http://www.lewenyixue.com/2024/08/03/Nat%20Methods%20%7C%20%E5%9F%BA%E4%BA%8E%E6%B5%8B%E5%BA%8F%E7%9A%841/
THE END