分高不卷,思路明显:聚焦难治性肿瘤
神经内分泌前列腺癌( NEPC )通常意味着严重的致死率和有限的治疗选择。 NEPC 细胞的准确鉴定对机制研究和临床应用都具有重要意义,但有效的 NEPC 生物标志物仍有待确定。今天分享一篇 2024 年 1 月 华中科技大学团队发表在 Theranostics ( IF : 12.4 ) 的分析文章。该研究为鉴定 NEPC 和 监测前列腺癌(PCa) 进展提供了一个有价值的工具。 
一研究背景
前列腺癌 (PCa) 是男性中第二常见的癌症,影响全球数百万男性。雄激素受体 (AR) 信号通路在前列腺癌的进展中起重要作用,而靶向 AR 信号通路可导致激素敏感性前列腺癌 (HSPC) 的反应。这种疾病经常复发成为一种更具侵袭性的表型,称为去势抵抗性前列腺癌 (CRPC) ,其中大多数仍然被组织学分类为腺癌 (CRPC-adeno) , AR 通路被重新激活。 其中,约 17%的 CRPC表现出不同程度的神经内分泌 (NE)表型 (CRPC-NE),并可能进一步发展为低分化 NE PCa (NEPC),这一亚型通常意味着严重的致死率和缺乏治疗选择。
随着患者接受多线治疗, NEPC 的发病率预计会增加。 NE 肿瘤细胞在组织学上可与复杂 PCa 微环境( TME )中的其他细胞区分开来;但它们的稀缺性,尤其是在疾病的早期阶段,导致早期 NEPC 经常被漏诊。 目前,NEPC的诊断主要依赖于几种生物标记物的免疫组化:阴性AR、高MKI67和阳性NE标记物。然而,这些蛋白在NE肿瘤细胞中的表达是异质性的,这大大降低了其诊断敏感性。
新一代测序技术的出现为全面描绘 NEPC 分子图谱提供了条件。 NEPC 的关键驱动因素已经确立,如原发性 PCa 中 FOXA1 和 SPOP 的突变、 RB1 缺失和 TP53 功能障碍诱导的系谱可塑性,以及晚期 PCa 中 EZH2 等多聚酶抑制复合体 -2 ( PRC2 )的激活。 同时,这些研究提出了10多个与NEPC相关的基因集,共包括数千个差异表达基因(DEGs)。然而,这些基因集具有相当大的异质性, 可能的原因包括: a. 这些研究大多基于数量有限的 NEPC 病例; b.CRPC-Adeno 和 NEPC 之间的基因表达谱惊人地相似; c. 这些基因集严重依赖于来自大块肿瘤而非 NE 肿瘤细胞的转录组数据。 因此,为了基础研究和临床转化的目的,仍然迫切需要开发敏感而特异的 NEPC 标志物。
在本研究中,作者首先收集了大量人类 PCa 的 scRNA-seq meta 图谱,发现已发表的 11 个 NE 基因集一致性差、作用力弱。 为了生成更好的NEPC预测因子,开发了一个综合管道,将bulk转录组数据、scRNA-seq数据和多种算法结合在一起,确定了771个高质量的NEPC特征标记和一个NE细胞内在基因特征,并构建了一个稳健的NEPC风险预测模型 。 通过使用来自人类 PCa 队列和 PCa 实验模型的大量数据集, NEPC 分类器在预测疾病进展为 NEPC 、预后和治疗反应性方面显示出卓越的能力,优于所有已发表的 PCa 预后模型。 NEPAL 模型为精确识别和描述 NE 肿瘤细胞提供了有用的参考。
二主要结果
1.以往的NEPC基因集一致性较低,检测效能较差
作者收集并分析了所有 11 个已发表的 NE 标记基因集,包括 9 个来自 bulk 转录组数据的 NEPC 基因列表、 1 个来自正常前列腺 scRNA-seq 的基因列表和 1 个来自 MSigDB 数据库的代表性泛前列腺肿瘤基因列表。 这11个基因集共包含1482个表达上调的NE标记(NEPC_Meta)。然而,这些基因集之间的重叠率很低,只有61个基因重叠了四次以上。
为了评估这些 NE 标记物的灵敏度和效率, 作者根据已发表的9个人类PCa scRNA-seq数据集(图1A),生成了一个全面的scRNA-seq参考图谱, 其中包含来自 66 个 PCa 肿瘤的 210,879 个单细胞,涵盖原发性 HSPC ( Pri )、 CRPC 、 mCRPC 和 NEPC 。通过相应的生物标记物共确定了 15 种细胞类型, 然后计算了每个样本中NE肿瘤细胞的频率(图1B)。 研究发现这些 NE 标志物( 894/1482 )中有一半以上并不只在 NE 肿瘤细胞或具有 NE 特征的患者中表达(图 1C )。至于上述 61 个重叠率较高的基因,尽管它们能很好地鉴别 NEPC 肿瘤,但一半以上( 41/61 )的表达丰度较低(在所有 NE 肿瘤细胞中的表达百分比低于 20% )(图 1D ),这意味着它们的效率较低。 最后,通过使用AUCell富集分析计算各基因集的NE评分,结果证实了大多数基因集在scRNA-seq数据中识别NE肿瘤细胞的特异性较低(图1E)。 这些结果表明,已发表的 NE 基因集的一致性较低,识别能力较差。
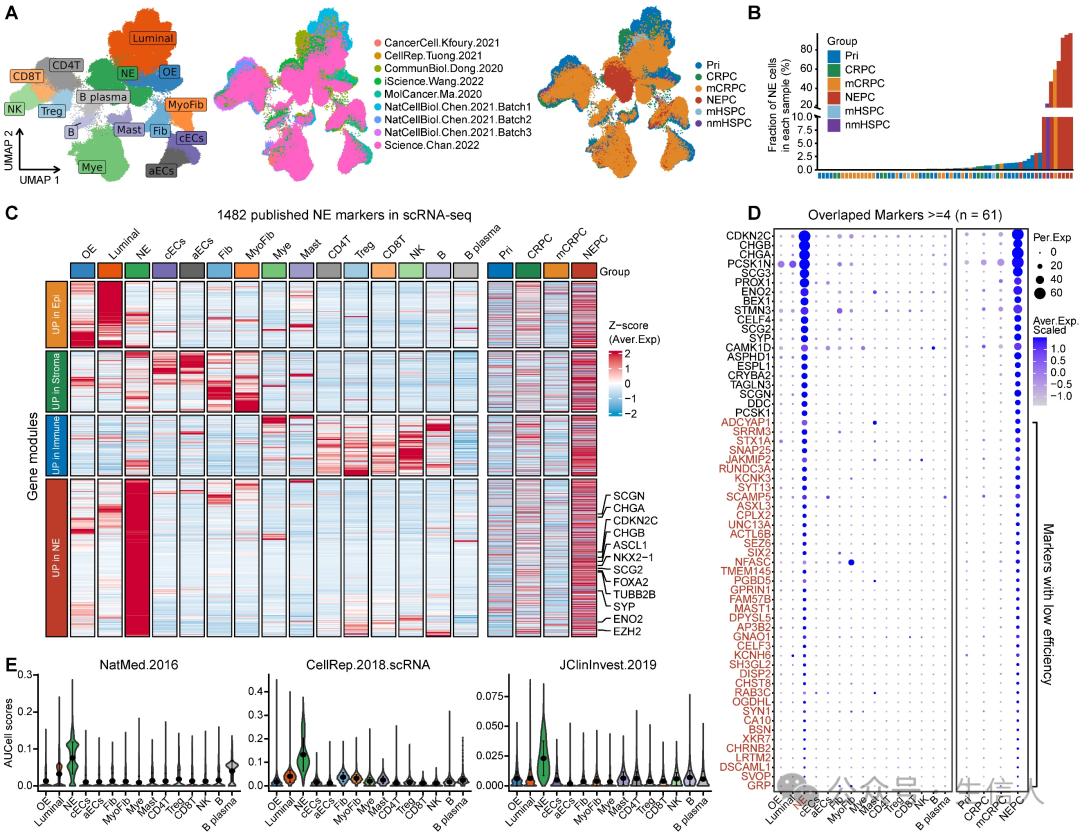 图 1. scRNA-seq 分析显示已发表的 NEPC 基因集合灵敏度低、效率低
图 1. scRNA-seq 分析显示已发表的 NEPC 基因集合灵敏度低、效率低
2.基于scRNA-seq和bulk RNA-seq meta数据库构建NEPC分类器
为了鉴定高质量的NEPC特征标记,作者设计了一个包括上述已发表的NEPC_Meta标记、基于bulk RNA-seq的PCa WGCNA基因模块和本研究PCa scRNA-seq meta图谱的计算管道(图 2A)。 最后,确定了587 个上调和 184 个下调的 NEPC 特征基因,统称为 NE_FG (图 2B )。由于侧重于癌细胞固有基因表达的特征被认为在临床上更有用, 作者 还将NE_FG 与 NE 肿瘤细胞的 DEGs 重叠,得到了两个 NE 细胞固有基因特征,分别称为 NE_UP ( n = 90 )和 NE_DN ( n = 40 )(图 2C )。所有NE_UP 特征基因都具有较高的表达丰度(所有 NE 肿瘤细胞的表达百分比均大于 20% )。
 图2. 基于scRNA-seq和bulk RNA-seq meta数据库,结合多种策略鉴定NEPC标记
图2. 基于scRNA-seq和bulk RNA-seq meta数据库,结合多种策略鉴定NEPC标记
为了进一步构建NEPC预测模型,作者对训练集应用了7种基于NE_FG的经典机器学习算法。此外,还基于ssGSEA算法构建了结合NE_UP和NE_DN的NE_UP_DN模型。 随后,利用这些NEPC 预测因子计算了 6 个 NEPC 肿瘤队列中每个样本的 NEPC 风险评分。在评价指标方面, 作者 计算了每种算法的平均C 指数(图 3A )和 R2 。在这些模型中, NE_UP_DN_ssGSEA 、 Enet [α= 0.01] 和 NE_UP_ssGSEA 排在前三位,它们的 ROC 曲线下面积也很高( AUC > 0.90 ,图 3B )。此外,除 RSF 和 GBM 模型外,大多数预测因子与 NEPC 预测得分的皮尔逊相关系数都很高(图 3C )。 根据 scRNA-seq meta 图谱,大多数算法在预测NEPC 风险评分与 NE 肿瘤细胞的细胞分数之间显示出较高的相关 性 (图3D )。 作者还通过六个验证数据集计算 AUC指数,将该模型与已发表的11个NEPC_Meta基因集进行了比较,模型一致优于之前的 NE基因列表(图3E)。
为了进行验证,作者选择了最佳分类器NE_UP_DN signature,并在scRNA-seq meta图谱和另外三个scRNA-seq验证集和空间转录组数据集中评估了其预测性能: a. 基于Smart-seq2 的 scRNA-seq 数据集; b. 基于荧光激活细胞分选( FACS )的单细胞数据集; c. de novo NEPC 与 HSPC 共存的空间基因表达图谱。结果显示, NE_UP_DN 与 AUCell 算法在所有验证集中都能精确预测 NEPC 细胞状态(图 3F-G )。 综上所述,本文的模型可以根据bulk和单细胞来源的转录组数据,稳健地区分具有NE特征的肿瘤。之后作者使用NE_UP_DN特征进行了后续分析,以下称其为NEPC算法(NEPAL)。
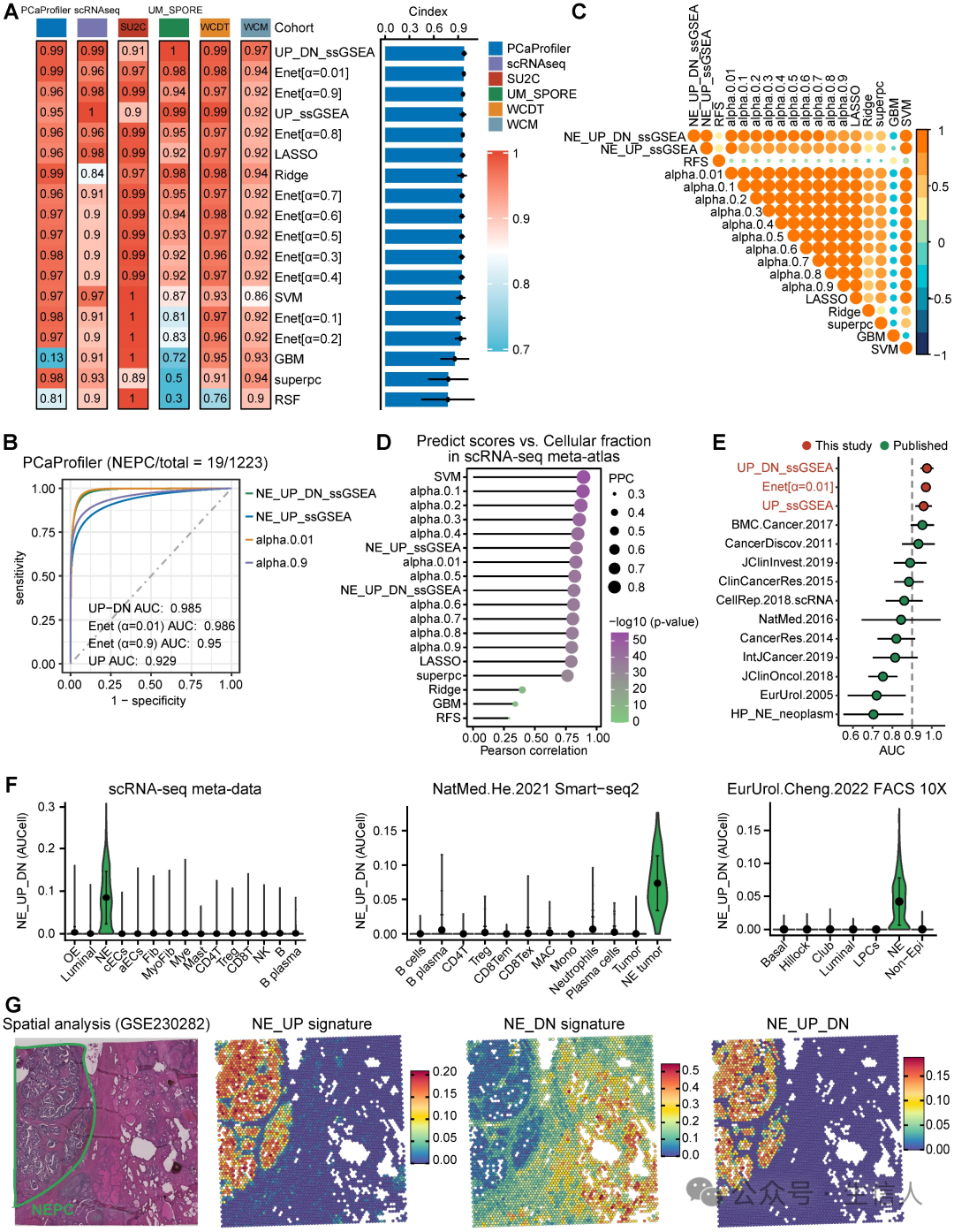 图3. 人工智能开发NEPC风险预测模型的构建与验证
图3. 人工智能开发NEPC风险预测模型的构建与验证
3.利用NEPAL来描绘PCa进展的路径
除了区分NEPC外,作者还假设NEPAL可以量化NEPC的进展,因为它将上调和下调的NE细胞内在特征基因都纳入了模型。为了评估这一假设,作者首先对scRNA-seq meta图谱中的21,526个NE肿瘤细胞进行了重新聚类,从而确定了8个NEPC亚群(图4A-B)。 这些亚群中 CHGA 、 SYP 、 ENO2 和 NCAM1 等经典的NE 标记物均呈异质性表达(图 4C )。采用 AUCell 算法的 NEPAL 在所有 NEPC 亚簇中几乎都有表达(图 4D )。接下来,对 8 NEPC 亚簇进行了伪时间和 CytoTRACE 分析(图 4E-F ),其进化轨迹与NEPAL 风险评分高度相关(图4G ),表明其在预测 NEPC 进展方面的实用性。 作者还 发现PCa 表达谱 中的NEPAL 风险指数与伪时间评分之间存在显著的相关性 ( 图 4h) 。此外, 作者 在TCGA-PRAD 、 CamCap 、 ICGC-PRAD 和 CPGEA 四个独立数据集中检查了尼泊尔风险指数与 Gleason 评分之间的关系。 总的来说,这些结果表明NEPAL可以用于预测NE前列腺癌的疾病进展。
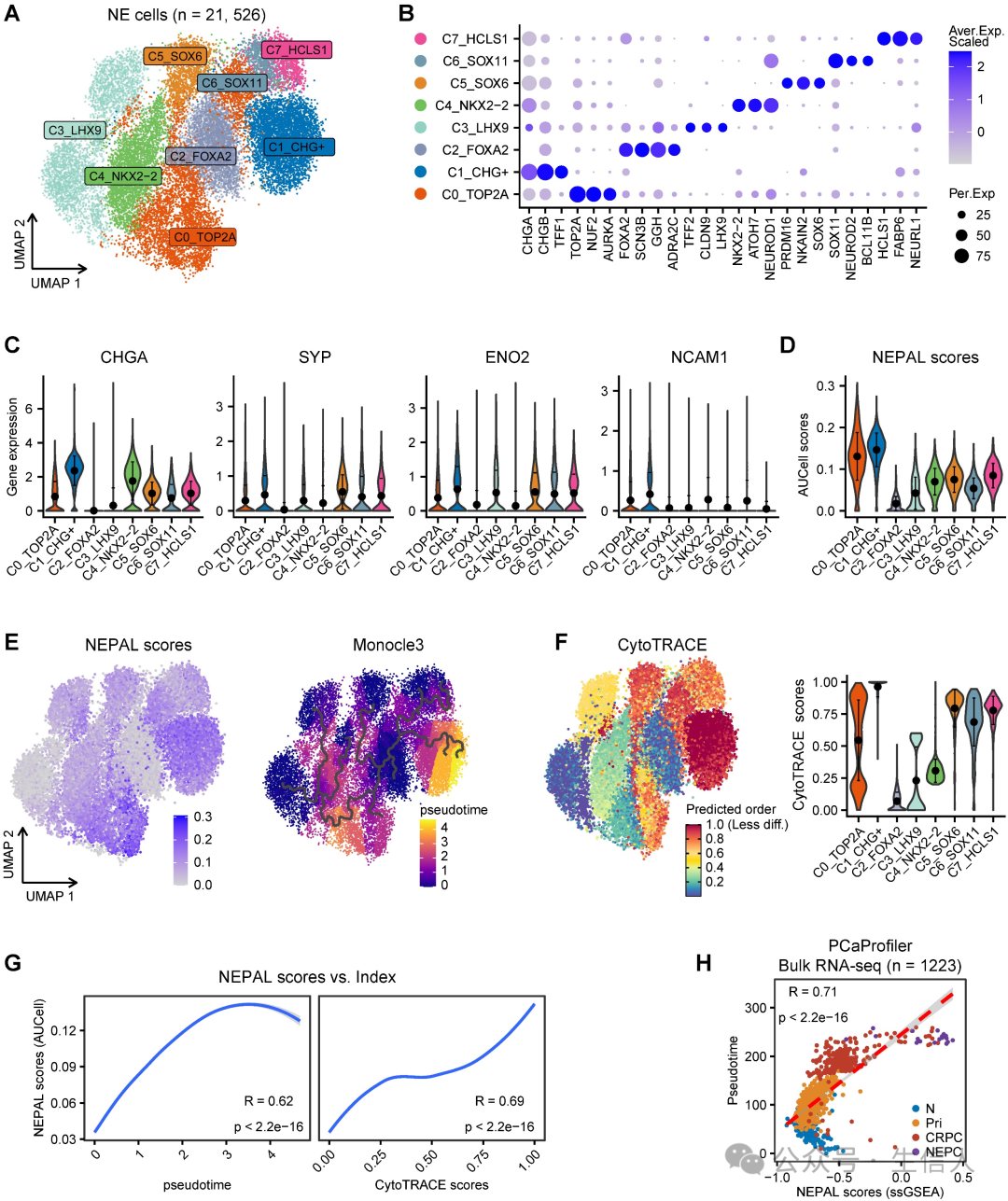 图4. NEPAL描绘PCa进展的路径
图4. NEPAL描绘PCa进展的路径
4.NEPAL在PCa实验模型中的应用
为了验证其实用性,作者进一步将NEPAL应用于PCa实验模型的转录组图谱。 对于来自CCLE 的 8 个人类 PCa 细胞系, NEPAL 准确地 为 NEPC 细胞系 NCHI-H660 分配了最高的 NEPC 风险分值。 DU145 、 22RV1 和 PC3 等 CRPC 细胞系紧随其后,而 MDA-PCa-2B 和 LNCaP 等激素依赖性细胞系的 NEPC 风险得分最低。此外, 作者 还观察到NEPAL 评分与 CHGA 和 SYP 等 NE 经典标记物之间存在较高的皮尔逊相关系数。
同时,作者将NEPAL应用于从2个人类 PCa PDX肿瘤和3个PCa转基因小鼠模型中生成的bulk转录组数据集。 PDX 数据库( UW/RA )包含 128 个人类 PCa 肿瘤的转录组数据,其中包括 87 个 CRPC 和 41 个 PDX 肿瘤。 在PDX肿瘤中,NEPAL评分与AR/NE状态的演变密切相关(图5A)。 同时,还观察到预测的NEPC 风险评分与 NE 标记物之间存在较高的皮尔逊相关系数(图 5B )。在一个独立的 PDX 队列中也多次检测到类似的关联(图 5C-D )。这一现象并不局限于人类 PCa , NEPAL 在两个小鼠 PCa 数据集中也显示出了一致的表现(图 5E )。最后, 根据 小鼠PCa 模型 RNA-seq 数据集, NEPAL 在预测 NEPC 状态方面再次表现出卓越的准确性, 表现出 显著的生存分层(图5F )。 这些结果增强了NEPAL鉴别NEPC的能力。
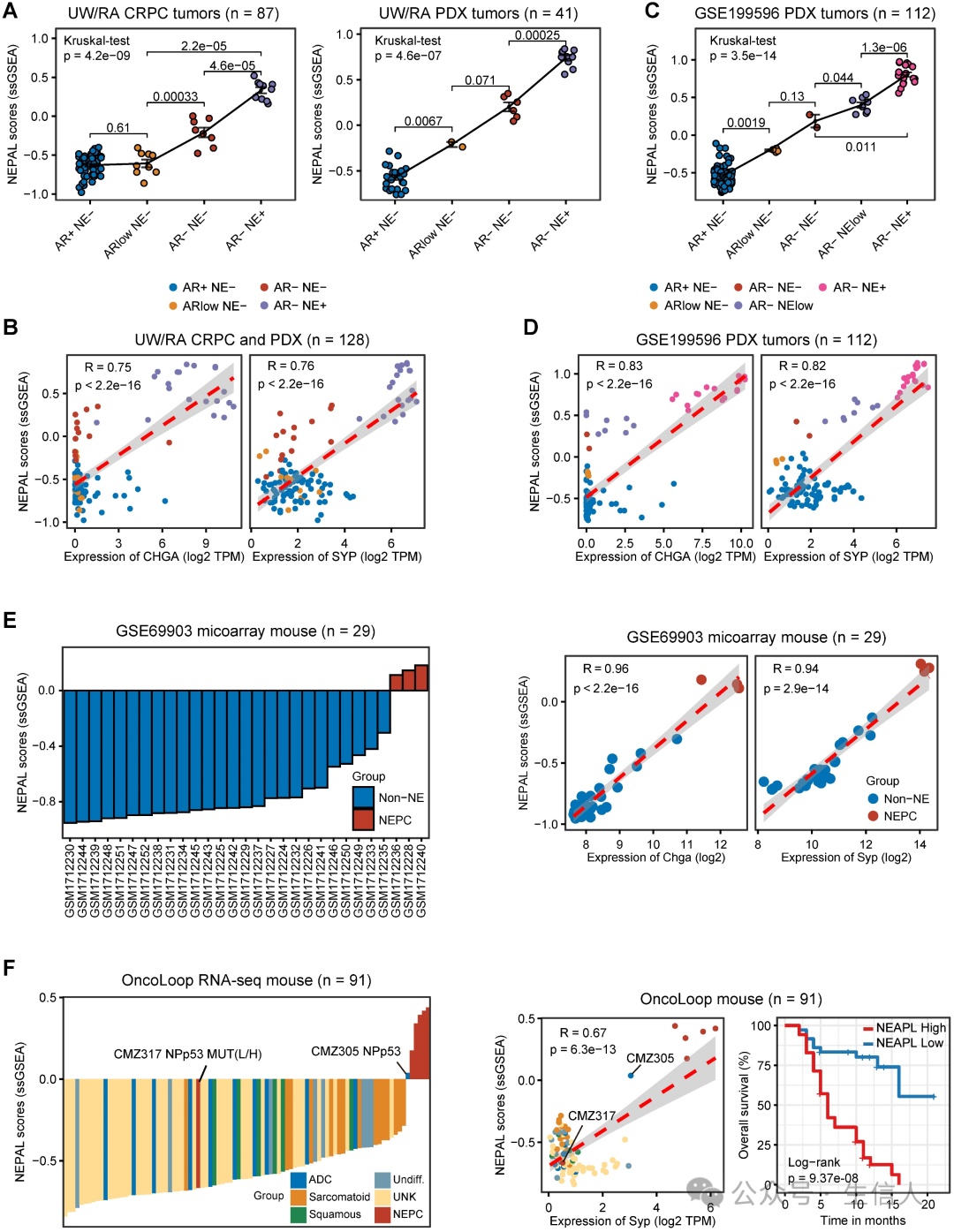
5.NEPAL的预后价值和生物学相关性
为了评估NEPAL模型的预后价值,作者收集了12个独立的bulk转录组数据集,包含2000多个人类PCa样本,其中10个数据集具有可用的预后信息。作者观察到NEPAL能有效地对HSPC的生化复发(BCR)和晚期PCa患者的总生存期(OS)进行分类(图6A-C)。 此外,根据现有的治疗信息,包括激素治疗、化疗和第二代AR 信号抑制( ARSI ), NEPAL 还能可靠地预测化疗和 ARSI 的耐药性。在 SU2C 、 UM/SPORE 、 MCTP 或 CPGEA 队列中,未接受过治疗组和接受过治疗组的 NEPAL 评分无明显差异,这表明患者之前的治疗史对 NEPAL 模型的预后准确性影响不大。 为了进一步比较NEPAL模型的预后能力,作者收集了20个已发表的预后模型,这些模型均由不同的机器学习算法生成,同时还包括传统的临床参数,如PSA评分、Gleason评分和肿瘤分期。 C 指数显示,在 10 个多中心 PCa 队列中, NEPAL 是比其他模型和传统临床参数最强大的特征(图 6D ),揭示了 NEPAL 模型在预后预测中的稳健性。
同时,在所有数据集中,NEPAL 风险评分与血统可塑性相关通路(如 EZH2、SOX2、NE 分化)的活性以及 RB1、PTEN 和 TP53 信号的缺失明显相关(图 6E)。 此外,NEPAL 风险评分还与晚期 PCa 的几个特征,即 AR-V 、细胞周期进展、 MYC 靶点、增殖和干性显著相关,而与雄激素反应和管腔特征则相反(图 6E )。 这些结果共同增强了NEPAL预测PCa患者预后、治疗反应性和分子特征的能力。
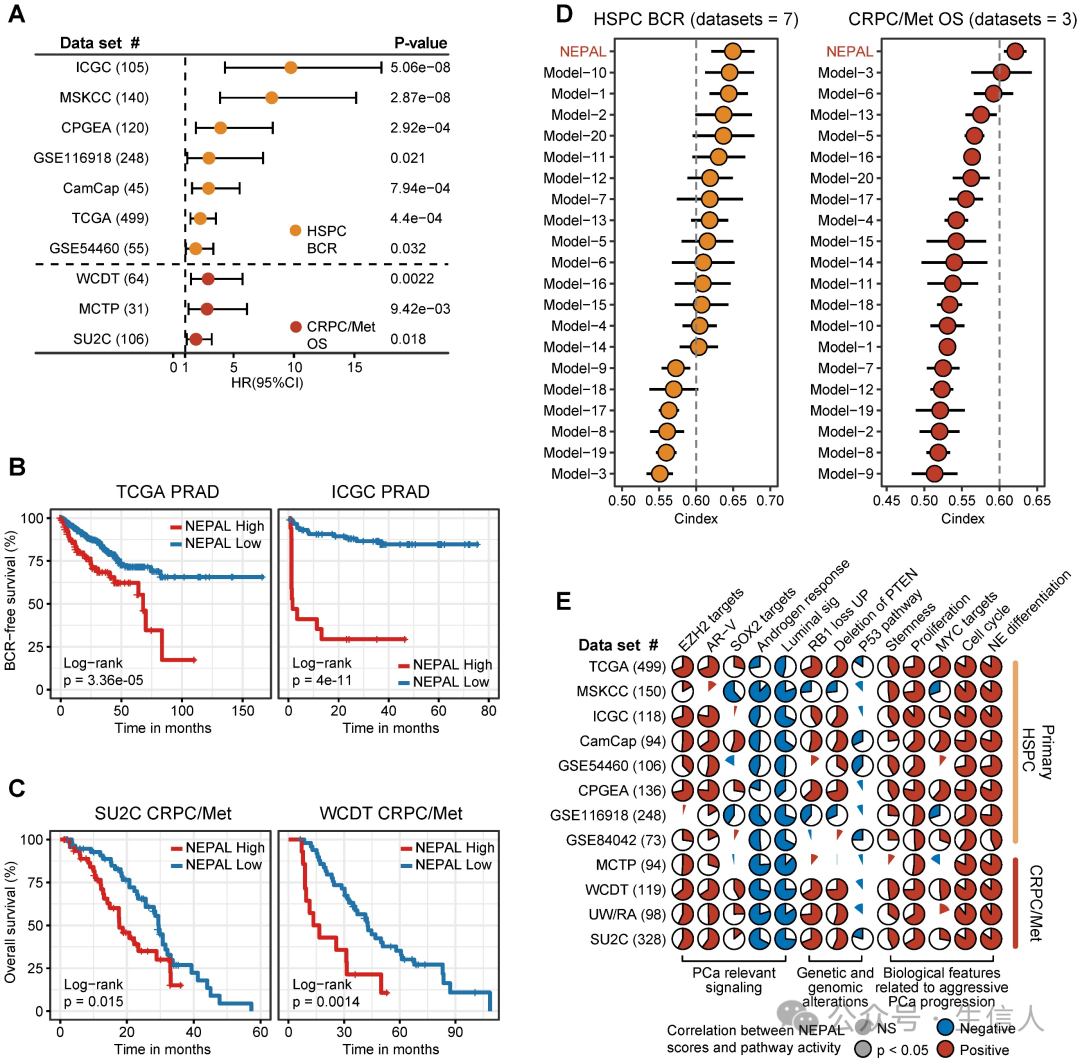 图6.人类PCa数据库中NEPAL的预后和分子特征
图6.人类PCa数据库中NEPAL的预后和分子特征
6.TME成分、患者参数和肿瘤分期对NEPAL预测准确性的影响
为了评估TME成分、患者年龄和种族以及肿瘤分期对NEPAL模型预测准确性的潜在偏差,作者对这些因素进行了分层分析。 结果显示,NEPAL 模型在不同的 TME 组别中对患者预后和 NEPC 风险保持了稳健的预测能力。同时, NEPAL 模型还能有效区分不同亚型 PCa 中具有 NE 特征的肿瘤。此外,基于患者年龄、种族和癌症分期的分层分析表明, NEPAL 模型对患者预后的预测能力不受病理因素的影响。 这些结果增强了NEPAL模型在预测NEPC风险和进展方面的普适性和有效性。
7.NEPAL揭示了NEPC的非遗传驱动因素
NEPC的发生和进展既有遗传因素,也有非遗传因素。作者根据NEPAL评分对TCGA PRAD和 SU2C CRPC/Met队列中的肿瘤进行了分层,并分析了它们的表达谱和体细胞突变。 在PCa 中最常突变的基因中,只有 TP53 在 PRAD 和 CRPC/Met 队列的 NEPC 高危组中比低危组显示出更高的突变率,而 AR 和 RB1 仅在 SU2C CRPC/Met 数据集中的 NEPC 高危组中观察到更高的突变率。此外,在 TCGA PRAD 数据集中,肿瘤突变负荷( TMB )和所有基因的突变计数都与 NEPC 风险评分显著相关,而在 SU2C CRPC/Met 数据集中则不显著。 接下来,作者评估了PCaProfiler中1223个组织(包括正常前列腺、原发性PCa、CRPC/Met和NEPC)的基因表达与NEPC风险评分的相关性(图7A)。 编码染色质重塑因子的关键基因,包括DNA 甲基转移酶( DNMTs )以及多聚酶抑制复合体 -2 ( PRC2 )的成员出现在了前列。对 TCGA PRAD 和 SU2C CRPC/Met 数据集的相同分析也反映了这一观察结果, 支持了表观遗传调节因子在 NEPC 中的关键作用。
此外,基于相关秩的GSEA 分析表明, NE 分化、胶质母细胞瘤( GB )可塑性、 PTEN 缺失、 EZH2 信号传导、 RB1 和 TP53 双敲除上调信号传导( LNCaP_DKO_UP )等与细胞系可塑性相关的通路,以及 E2F 靶点、 G2M 检查点和 MYC 信号传导等与增殖和干性相关的通路是最显著激活的通路。另一方面,与 HSPC 相关的通路,如雄激素反应、 IRE1α-XBP1s 信号传导、 SPOP 缺失和 AR 信号传导则受到抑制(图 7B )。 最后,结合VIPER方法的TF活性推断,作者分别描绘了AR信号、P53和RB1通路以及表观遗传调控等四种与NE转分化相关的通路的信号网络(图7C)。 此外,NEAPL 结合 VIPER 算法鉴定出了与 NEPC 相关的先驱 TFs ,包括以前建立的 TFs ,如 FOXA2 、 ASCL1 和 MYCN ,以及新型 TFs ,如 XBP1s 、 PHTF 、 LHX2 和 NANOS1 。 这些TFs是否以及如何单独或合作驱动NEPC的进展将是未来研究的兴趣所在。
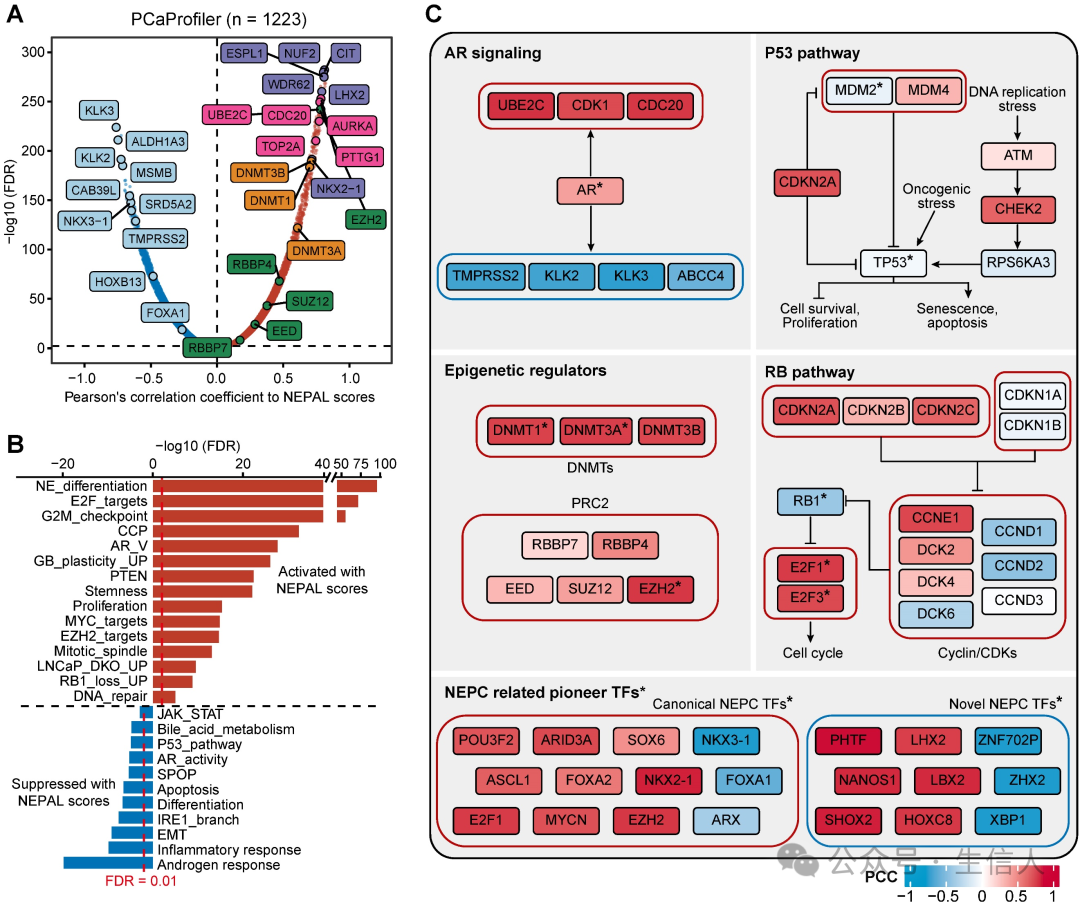 图7.NEPAL对NEPC非遗传进化驱动基因的预测
图7.NEPAL对NEPC非遗传进化驱动基因的预测
8.NEPAL: 使用转录组数据预测NEPC风险评分的计算框架
为了方便用户应用,作者 推出了一个R 软件包 NEPAL ,它集成了本研究中测试的已发表 NE 基因组、用于批量转录组数据的 ssGSEA 算法、用于 scRNA-seq 数据的 AUCell 算法、多种机器学习模型和数据可视化。此外 , NEPAL 还支持将小鼠和人类转录组数据作为输入。该工作流 程 与Seurat scRNA-seq 工具包具有良好的兼容性,可广泛适用于不同平台的 scRNA-seq 数据集 。
NEPAL的R软件包现已在Github上发布(https://github.com/Famingzhao/NEPAL)
三总结
这项工作构建并广泛验证了稳健的 NEPC 风险预测模型 NEPAL ,该模型可用于基础研究目的的精准识别 NEPC ,并为未来临床转化奠定了坚实的基础。
参考文献
Zhang T, Zhao F, Lin Y, Liu M, Zhou H, Cui F, Jin Y, Chen L, Sheng X. Integrated analysis of single-cell and bulk transcriptomics develops a robust neuroendocrine cell-intrinsic signature to predict prostate cancer progression. Theranostics. 2024 Jan 1;14(3):1065-1080. doi: 10.7150/thno.92336. PMID: 38250042; PMCID: PMC10797290.
链接:http://www.lewenyixue.com/2024/06/26/%E5%88%86%E9%AB%98%E4%B8%8D%E5%8D%B7%EF%BC%8C%E6%80%9D%E8%B7%AF%E6%98%8E%E6%98%BE%EF%BC%9A%E8%81%9A%E7%84%A6%E9%9A%BE%E6%B2%BB%E6%80%A7%E8%82%BF%E7%98%A4/