Nature | 研究揭示髓母细胞瘤常见突变的致癌机制
理解突变如何导致蛋白质-蛋白质相互作用的改变,以及这些改变如何驱动肿瘤的发生,仍癌症研究一个重要的未解决问题。KBTBD4是一种CULLIN3-RING E3泛素连接酶复合物的底物受体,其突变在髓母细胞瘤中频繁出现,而这些突变使KBTBD4 获得功能,从而诱导转录辅阻遏物CoREST5的异常降解。
近日, Brian B. Liau 和合作伙伴们在 Nature 上发表题为 Converging mechanism of UM171 and KBTBD4 neomorphic cancer mutations 文章, 揭示了功能获得性E3突变和分子胶降解剂UM171之间基于形状互补性的吻合机制,证明HDAC1/2抑制剂可以阻断突变型KBTBD4–HDAC1界面和KBTBD4突变型髓母细胞瘤细胞的增殖。

在泛素-蛋白酶体系统中,人类疾病突变早已知道会损害几种E3泛素连接酶的功能。相比之下,促进底物蛋白异常降解的E3功能获得性突变最近被发现。尽管有通过改变连接酶稳定性或调节性导致非计划的底物泛素化的多形态(hypermorphic)E3突变病例,但直接诱导新底物参与和降解的E3突变,即“新降解”,是E3连接酶失调的新例子。
KBTBD4(一种 CULLIN3-RING E3 连接酶的底物受体)的癌症突变主要发生在 KELCH 重复 β 螺旋桨 2b-2c 环的热点中,具有相当大的分子多样性,包括1-5 个氨基酸的插入或缺失(insertion or deletion)以及点替换。值得注意的是,KBTBD4 还参与了 UM171 的机制,后者是一种诱导 CoREST 降解的造血干细胞扩增小分子激动剂。为揭示UM171 和 KBTBD4 癌症突变之间惊人的相似机制, 该团队做了几个关键实验:
-
深度突变扫描 :绘制了KBTBD4 癌症热点的突变图,揭示了插入和替换可以促进功能获得的不同偏好以及热点相互作用中有关的关键残基。KBTBD4的癌症突变通过直接靶向HDAC1/2,促进CoREST(一种控制染色质可及性和转录的复合物)的降解,这种降解会改变表观遗传程序,进而改变转录程序,促进癌细胞干细胞的增加,从而驱动肿瘤发生。
-
冷冻电子显微镜分析 :对与 LSD1–HDAC1–CoREST (LHC 综合体)结合的两个不同 KBTBD4 癌症突变体的低温电子显微镜分析表明,KBTBD4 同型二聚体与 HDAC1 不对称地结合,具有两个 KELCH 重复的β-螺旋结构域。髓母细胞瘤突变稳定了 HDAC1 和其中一个 KBTBD4 β-螺旋之间的界面,使一个庞大的侧链插入 HDAC1 活性位点。
最近的研究表明,KBTBD4作用机制与UM171 相吻合, UM171 通过促进 CoREST 的泛素化和降解在表型上和髓母细胞瘤中KBTBD4突变相似。在相关的研究中,研究人员确定UM171的加入稳定了KBTBD4-LHC 复合物的结构,并证明了UM171充当分子胶以诱导 E3 和 HDAC1 之间的复合物形成。比较KBTBD4-UM171-LHC 复合物与两个和LHC 结合的 KBTBD4突变体之间的结构,明显看出两机制的吻合,如图1所示。通过该机制,分子胶和癌症突变可以补充和优化 E3 连接酶和 HDAC1 之间较差的蛋白质-蛋白质界面,从而驱动它们的结合。
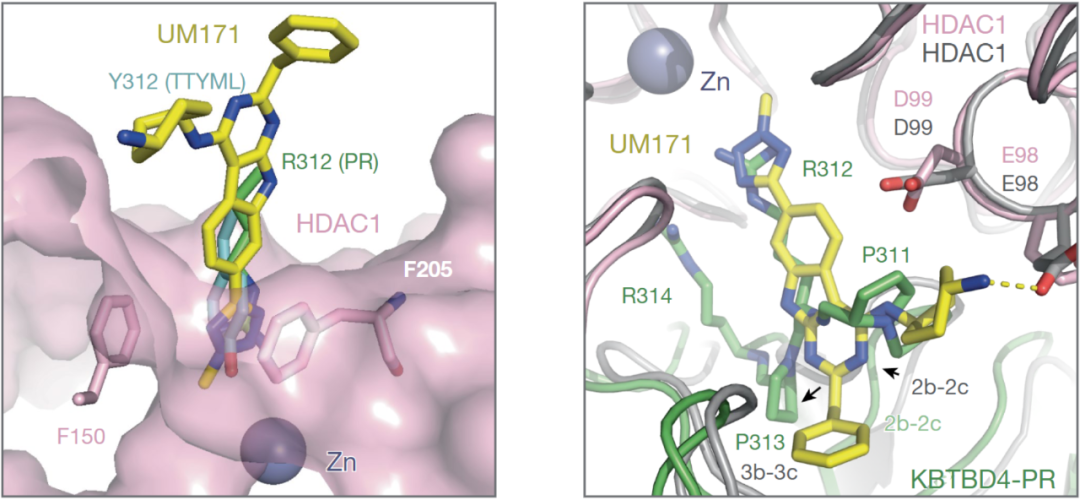
图1,左-UM171(黄色和蓝色棒)、突变型KBTBD4- Tyr312 侧链(青色和红色棒)以及另一突变型 KBTBD4- Arg312 侧链(绿色和蓝色棒)在 HDAC1 活性位点口袋处(粉色)的比较。深紫色棒显示了两个苯丙氨酸残基,勾勒出 HDAC1 活性位点通道的入口。右- UM171(黄色和蓝色棒状图)与 突变型KBTBD4的 2b-2c 环状图的比较,其中 KBTBD4–UM171–HDAC1 结构通过 HDAC1 与突变型 KBTBD4–HDAC1 结构叠加,界面处关键残基的侧链以红色棒状标示。
-
药物干预实验 :结构和深度突变扫描结果表明KBTBD4 MB 突变体通过去乙酰化酶活性位点的热点相互作用直接与 HDAC1/2 结合,从而促进 CoREST 降解。这些观察结果意味着HDAC1/2 活性位点抑制剂可以在空间上阻断突变连接酶的结合,从而阻断其致癌功能。突变型KBTBD4-HDAC1 与与辛二酰苯胺羟肟酸 (SAHA) 结合的 HDAC2 的叠加分析支持这一观点,即 HDAC1/2 抑制剂和插入的精氨酸残基可能会发生位置冲突。用SAHA,CI-994(一种 2-氨基苯甲酰胺衍生的抑制剂)和RBC1HI(新型 HDAC1/HDAC2 抑制剂,Regenacy Brain I 类 HDAC 抑制剂)进行试验, 证明了这三种抑制剂可以保全由两种突变KBTBD4-P 和 KBTBD4-PR 表达引起的 CoREST 降解, 如图2所示。
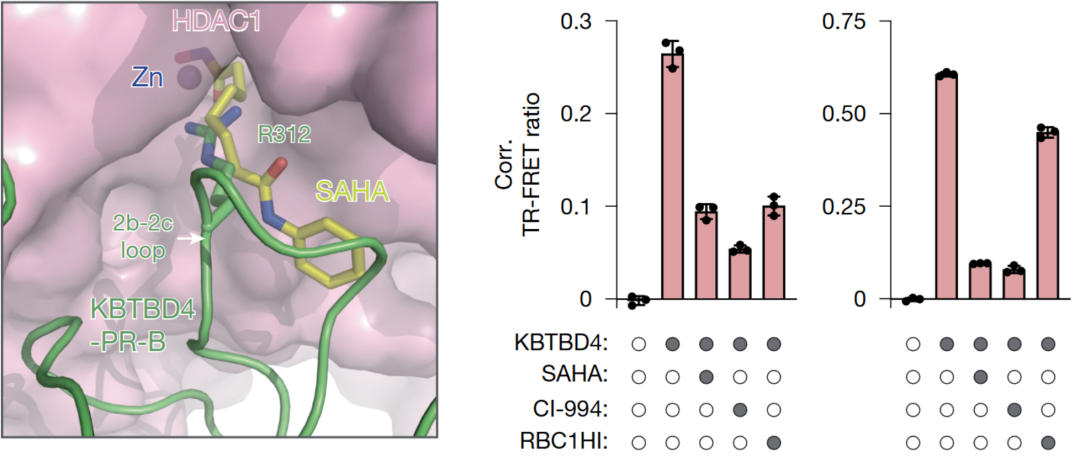
图2,左-SAHA(黄色)与 突变型KBTBD4 2b-2c 环(绿色)中央精氨酸残基之间的位置冲突。右-细胞增殖实验, DMSO,SAHA,CI-994或 RBC1HI与带有标示的KBTBD4 突变体之间的荧光素信号(纵轴),两次生物学重复
了解突变是如何实现新蛋白质-蛋白质相互作用(PPI)的分子机制不仅对于理解疾病病因至关重要,而且对于指导治疗方法(例如分子胶和 PPI 抑制剂)用化学调节这些界面也至关重要。该研究表明, KBTBD4的癌症突变通过与HDAC1的相互作用促进CoREST的降解,且可以通过小分子药物进行调节,这为针对KBTBD4突变的治疗提供了新的思路。
未来的研究应集中在扩展对其他相关突变的研究,探索其在不同类型癌症中的作用;开发针对KBTBD4突变的特异性治疗策略,例如分子胶UM171和HDAC1/2抑制剂;进一步研究KBTBD4与其他蛋白质的相互作用,以全面理解其在肿瘤发生中的角色。
撰文

责编

制作
排版 | 车洁 校对 | uu
▼滑动查看更多▼
Cancer mutations can create neomorphic protein-protein interactions to drive aberrant function1,2. As a substrate receptor of the CULLIN3-RING E3 ubiquitin ligase complex, KBTBD4 is recurrently mutated in medulloblastoma3, the most common embryonal brain tumour in children4. These mutations impart gain-of-function to KBTBD4 to induce aberrant degradation of the transcriptional corepressor CoREST5. However, their mechanism remains unresolved. Here we establish that KBTBD4 mutations promote CoREST degradation through engaging HDAC1/2 as the direct target of the mutant substrate receptor. Using deep mutational scanning, we chart the mutational landscape of the KBTBD4 cancer hotspot, revealing distinct preferences by which insertions and substitutions can promote gain-of-function and the critical residues involved in the hotspot interaction. Cryo-electron microscopy analysis of two distinct KBTBD4 cancer mutants bound to LSD1-HDAC1-CoREST reveals that a KBTBD4 homodimer asymmetrically engages HDAC1 with two KELCH-repeat β-propeller domains. The interface between HDAC1 and one of the KBTBD4 β-propellers is stabilized by the medulloblastoma mutations, which insert a bulky side chain into the HDAC1 active site pocket. Our structural and mutational analyses inform how this hotspot E3-neosubstrate interface can be chemically modulated. First, we unveil a converging shape-complementarity-based mechanism between gain-of-function E3 mutations and a molecular glue degrader, UM171. Second, we demonstrate that HDAC1/2 inhibitors can block the mutant KBTBD4-HDAC1 interface and proliferation of KBTBD4-mutant medulloblastoma cells. Altogether, our work reveals the structural and mechanistic basis of cancer mutation-driven neomorphic protein-protein interactions.
DOI: 10.1038/s41586-024-08533-3
版权声明:本文为“乐问号”作者或机构在乐问医学上传并发布,仅代表该作者或机构观点,不代表乐问医学的观点或立场,不能作为个体诊疗依据,如有不适,请结合自身情况寻求医生的针对性治疗。
链接:http://www.lewenyixue.com/2025/03/27/Nature%20%7C%20%E7%A0%94%E7%A9%B6%E6%8F%AD%E7%A4%BA%E9%AB%93%E6%AF%8D%E7%BB%86%E8%83%9E%E7%98%A4%E5%B8%B8%E8%A7%81/
THE END





赶快来坐沙发