



靶向RAS–ERK信号通路的癌症治疗
前言
RAS-RAF-MEK-ERK (简称 ERK 信号通路)是调控细胞增殖与存活的核心通路,其异常活化与多种癌症的发生发展密切相关。在生理条件下,该通路通过多级反馈调控维持稳态,但在约三分之一的实体瘤中检测到 RAS 突变(如 KRAS 、 NRAS ),约 8% 的肿瘤携带 BRAF 突变(如 BRAFV600E ),导致通路持续激活并驱动肿瘤恶性转化。尽管 RAF 抑制剂(如维莫非尼、达拉非尼)和 MEK 抑制剂(曲美替尼)已在 BRAF 突变型黑色素瘤中展现显著疗效,但耐药问题普遍存在,且 RAS 突变型肿瘤的靶向治疗仍充满挑战。
目前,针对该通路的药物开发已形成多层次策略:靶向上游 RAS 的激活机制、抑制中游 RAF/MEK/ERK 激酶活性,或通过联合用药阻断反馈激活。新一代 RAF/MEK 抑制剂、首款 ERK 抑制剂,以及针对 RAS 的直接靶向策略陆续进入临床试验,为突破耐药瓶颈和扩展适应症带来新希望。
- 02 -
RAS-RAF-MEK-ERK信号通路
ERK 信号通路的核心是一条高度保守的激酶级联反应: 细胞外生长因子与膜受体结合后,激活 RAS 蛋白( KRAS 、 NRAS 或 HRAS ),诱导其从非活性的 GDP 结合态转为活性的 GTP 结合态。 活化的 RAS 招募 RAF 激酶至细胞膜并促使其二聚化,继磷酸化激活 MEK ,最终由 MEK 激活 ERK 激酶。 ERK 通过磷酸化转录因子(如 MYC 、 FOS )和细胞周期调控蛋白,驱动细胞增殖、存活、代谢重编程及转移。
该通路的病理激活主要源于 RAS 或 BRAF 突变(占人类肿瘤的 40% 以上)。 例如, KRAS G12 突变(占胰腺癌 90% )导致 GTP 酶活性丧失, BRAFV600E 突变(见于黑色素瘤)无需二聚化即可持续激活。 此外,表观遗传调控(如 NF1 基因缺失)或上游受体酪氨酸激酶( RTK )异常活化也可能增强通路信号。 值得注意的是, RAF 抑制剂的 “ 矛盾激活 ” 现象(在野生型 BRAF 细胞中反上调 ERK 信号)揭示了通路的复杂调控机制,进一步提示联合治疗的必要性。
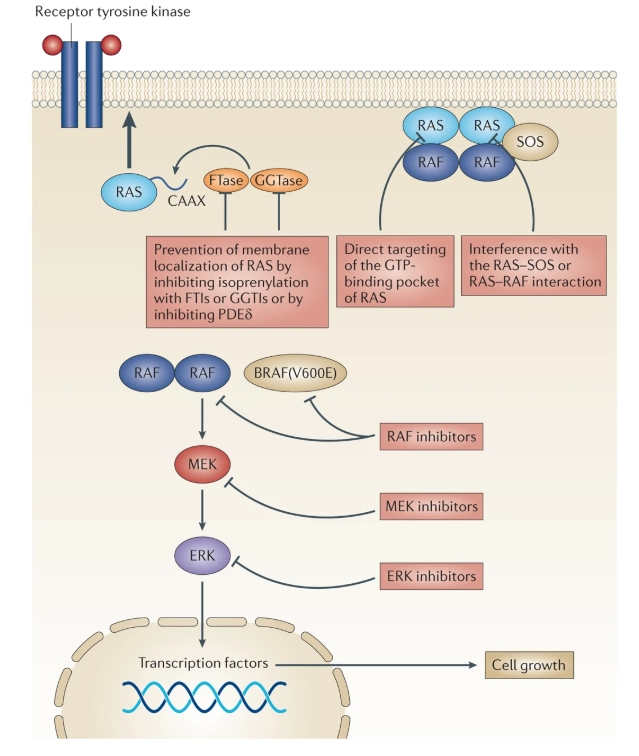
通过系统性解析,研究已明确该通路的双重角色: 生理性的精细调控维持正常组织稳态,而致癌性激活则赋予肿瘤恶性特征。 因此,精准选择抑制节点、平衡治疗效果与毒性成为开发策略的关键。
- 03 -
RAS作为信号通路上游的“分子开关”,其直接靶向开发曾被视为“不可成药”难题。传统策略聚焦间接干预:抑制RAS膜定位所需的法尼酰化修饰(如法尼酰转移酶抑制剂FTIs),但因RAS的“异构体逃逸”(如通过香叶酰修饰旁路激活)而失败。近年突破集中于两方面:其一是靶向RAS与鸟苷酸交换因子(如SOS)的相互作用,例如片段化合物通过结合RAS表面的浅凹槽阻断SOS激活(临床前阶段);其二是针对特定突变体(如KRAS G12C)的共价抑制剂Sotorasib(AMG510)和Adagrasib(MRTX849),通过“开关口袋”锁定突变型KRAS的失活构象,已在肺癌中获批,但胰腺癌等适应症仍需验证。
新兴策略包括靶向RAS伴侣蛋白PDEδ(调控KRAS亚细胞定位)、干预RAS下游效应通路(如RAF或PI3K双抑制改善RAS突变型肿瘤反应),以及RNA干扰技术。此外,结构生物学和人工智能驱动的新药设计(如PROTAC)加速了KRAS G12D/V等难靶突变体的抑制剂开发。尽管直接靶向RAS仍面临成药性挑战,但KRAS G12C突破性疗法的成功,为其他突变亚型提供了信心。
- 04 -
- 05 -
- 06 -
- 07 -
参考文献:
TargetingRAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov.2014Dec;13(12):928-42
链接:http://www.lewenyixue.com/2025/02/18/%E9%9D%B6%E5%90%91RAS%E2%80%93ERK%E4%BF%A1%E5%8F%B7%E9%80%9A%E8%B7%AF%E7%9A%84%E7%99%8C%E7%97%87%E6%B2%BB%E7%96%97/

赶快来坐沙发