Nature重磅:乳腺癌治疗新策略!选择性抑制AKT1,减少副作用
99%的 乳腺癌 发生在女性,男性仅占1%,名副其实的女性健康“第一杀手”, 世界卫生组织国际癌症研究机构 (IARC) 最新数据显示,2022年全球女性乳腺癌发病人数高达 231万 ,且发病率仍在持续上升。 面对这一严峻挑战,科研人员正在不断探索新的治疗策略。
PI3K/AKT/mTOR 是乳腺癌治疗研究中的核心信号通路,而AKT作为其中承前启后的关键节点,在细胞生长、增殖和新血管生成中发挥关键作用。 目前已有一款AKT选择性抑制剂 卡匹色替 (Capivasertib) 获得美国FDA批准上市,在国内还处于临床Ⅲ期阶段,有望在2025年上市。
AKT抑制剂主要结合在激酶活性位点或激酶域和PH结构域的变构口袋上。这些位点在AKT家族几乎完全相同,难以区分AKT1、AKT2、AKT3。研究显示,AKT抑制剂的高血糖的副作用归因于AKT2被抑制,因此, 迫切需要一种选择抑制AKT1而对AKT2影响较小的药物。
近日,加州大学旧金山分校的研究人员在国际顶尖学术期刊 Nature 上发表了题为: Mutant-selective AKT inhibition through lysine targeting and neo-zinc chelation 的研究论文。
该研究通过结构设计,开发出了 可逆的、选择性共价结合 AKT1(E17K)突变 的新型抑制剂,在细胞中实现持续的靶标结合,为 乳腺癌 提供了新的治疗策略。

蛋白激酶AKT也被称作蛋白激酶B (protein kinase B,PKB) ,属于丝氨酸/苏氨酸蛋白激酶,同时也是致癌基因。AKT位于PI3K-AKT-mTOR信号通路 (PAM) 的中心节点,前接磷脂酰肌醇3-激酶 (PI3K) ,后启鼎鼎大名的哺乳动物雷帕霉素靶蛋白mTOR激酶。PAM整合细胞外和细胞内信号,在细胞代谢、生长、增殖中发挥核心调控作用。正常细胞中AKT自身抑制,在癌症中因突变而发生信号上调,从而驱动细胞增殖,是药物开发的重要靶点。
AKT已发现三种亚型: AKT1 、 AKT2 、 AKT3 ,具有80%以上的同源性,仅在PH结构域和激酶结构域连接处存在较大差异,但是它们在体内分布和生理功能上并非完全相同。因此在药物开发过程中需要选择性靶向AKT不同亚型。
AKT抑制剂按照结合位点不同,可以分为PH结构域抑制剂、变构抑制剂和ATP竞争性抑制剂。例如 卡匹色替 (Capivasertib) 属于ATP竞争性抑制剂,同时抑制AKT三种亚型,与Faslodex (氟维司群) 联用,治疗HR + /HER2 - 的晚期或转移性乳腺癌成年患者。
高血糖的副作用是广谱AKT抑制剂的剂量限制性毒性。高血糖及其它副作用导致Capivasertib剂量减少,可能导致对AKT1(E17K)的抑制不足。小鼠和人类基因组研究显示,高血糖副作用归因于对AKT2的抑制。
AKT1 第17位的谷氨酸 替换为 赖氨酸,即 AKT1 (E17K) , 是 其 最常见的突变。 在这项最新研究中,研究团队针对该突变,设计了水杨醛抑制剂化合物1-3。 结果显示,化合物3在驻留时间和热稳定性方面对AKT1(E17K)选择性强于野生型AKT1和AKT2。 在癌细胞模型中,化合物3可通过抑制AKT信号通路传导,降低癌细胞活力。 水杨醛探针3-炔烃直接证明了在细胞中的共价结合。
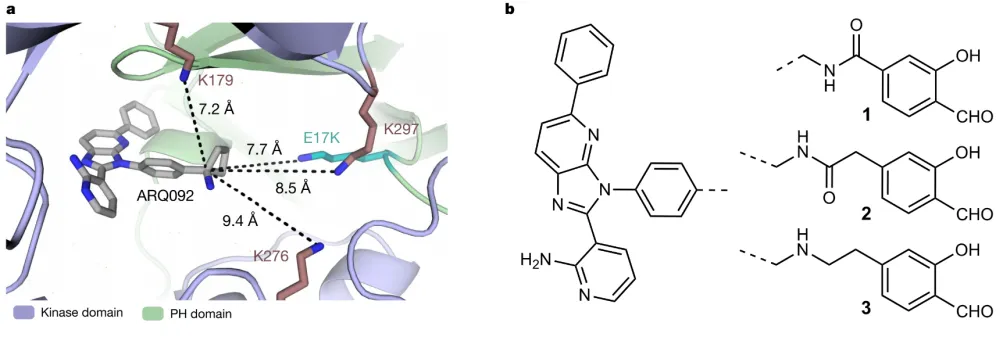
研究团队还开发了具有改善的药代动力学和选择性的氟化水杨醛化合物4,具有更好的药代动力学和结合停留时间。在荷瘤小鼠中,化合物4可以剂量依赖性的阻止肿瘤生长。与变构抑制剂ARQ092相比,化合物4不会影响小鼠自身体重和血糖水平。
总的来说,研究团队开发的共价抑制剂,能够特异性与 AKT1(E17K) 突变的赖氨酸结合,形成可逆的共价连接物。此连接物与内源性Zn 2+ 结合,进一步提供抑制剂与 AKT1(E17K) 是结合稳定性和选择性,进而避免因AKT2抑制而引发的高血糖副作用,为开发新的乳腺癌治疗策略提供坚实的科学基础。
深入研究AKT激酶信号机制、探索AKT激酶新的调节因子、开发更有效的AKT激酶抑制剂是未来的研究方向。AKT功能探索及应用依旧任重道远。 随着 SignalChem Biotech 的加入, 义翘神州 能够提供更广泛的激酶产品,具有高活性和高纯度,支持癌症和其他疾病中PI3K/AKT/mTOR信号通路的相关研究。
链接:http://www.lewenyixue.com/2024/11/28/Nature%E9%87%8D%E7%A3%85%EF%BC%9A%E4%B9%B3%E8%85%BA%E7%99%8C%E6%B2%BB%E7%96%97%E6%96%B0%E7%AD%96%E7%95%A5%EF%BC%81%E9%80%89%E6%8B%A9/