运用多组学方法表征肿瘤细胞的可塑性
今天小编分享的主题是 肿瘤细胞的可塑性 。细胞可塑性是指细胞被重新编程并改变其命运和身份的能力。细胞可塑性在某些情况下是有利的(如损伤后体内平衡恢复和组织再生),但在某些情况下是有害的(如肿瘤的转移)。
在肿瘤进展过程中,肿瘤细胞可以在不同的细胞状态之间切换,获得新的表型和功能特征以克服选择性压力,而这一过程主要由细胞可塑性介导。因此,细胞的可塑性在很大程度上促进了肿瘤内的异质性,增加了肿瘤细胞的适应性,并在很大程度上促进了肿瘤的生长、转移和对治疗的抵抗。
这次小编分享两篇综述。第一篇于 2023年 发表在 Nature Cancer (影响因子=23.4993),题目为“ Cancer cell plasticity during tumor progression, metastasis and response to therapy ” 。这篇文章回顾了驱动细胞可塑性,从而促进肿瘤的生长、增殖、转移和药物耐受的内在和外在机制,并且阐述了如何利用细胞可塑性进行抗肿瘤治疗。

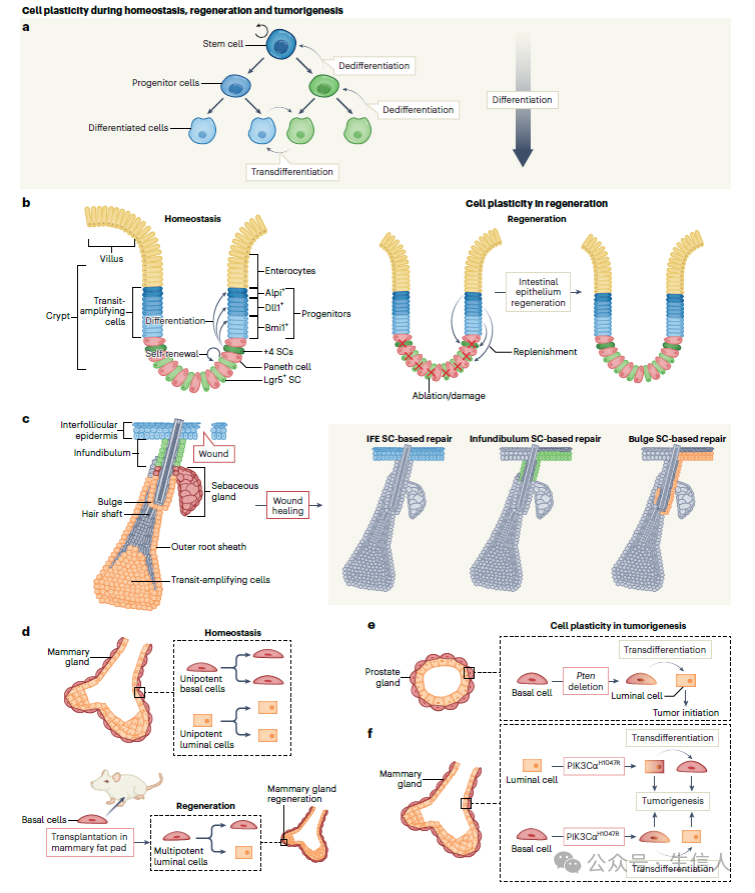 图1体内平衡、再生和肿瘤发生过程中的细胞可塑性
图1体内平衡、再生和肿瘤发生过程中的细胞可塑性 (a) 细胞可以通过去分化(分化细胞在同一谱系中恢复到未分化状态)、转分化(分化的细胞转化为另一个分化的细胞谱系,形成化生的基础)和上皮间质转化(EMT)即上皮细胞失去上皮特征(如细胞-细胞连接和极性)并获得间充质表型的过程,来显示可塑性。
(b) 肠上皮是哺乳动物中自我更新最快的组织之一。Lgr5是小肠和结肠中启动隐窝绒毛自组织小鼠类器官形成的干细胞的标志。肠隐窝含有干细胞和转运扩增祖细胞,它们可以在再生条件下恢复到多能状态。在小鼠中消除 Lgr5 + 干细胞谱系后,表达Bmi1的已分化细胞能够维持稳态并补充 Lgr5 + 干细胞池。甚至更多分化的 Alpi + 肠细胞祖细胞也可以恢复为 Lgr5 + 细胞。 损伤后,由 Lgr5 + 细胞衍生而来的前体,如分泌性 Dll1 + 祖细胞或Paneth细胞,可以恢复为 Lgr5 + 细胞,以补充干细胞池并使小鼠再生。
(c) 皮肤表皮由毛囊皮脂腺单位组成,其中包含一个毛囊,其相关的皮脂腺和周围的毛囊间表皮。在体内平衡过程中,这些不同的区域由各自的单能干细胞池维持。在伤口愈合过程中,不同的毛囊间表皮干细胞和祖细胞被招募。毛囊和毛囊干细胞向上迁移到毛囊间表皮,逐渐重编程为毛囊间表皮干细胞,增殖并有助于皮肤修复。该微环境对于这种重编程很重要:当小鼠毛囊干细胞被耗竭时,空的微环境可以招募更多已决定分化的细胞,使其逆转为干细胞样状态并稳定地补充干细胞池。
(d) 许多腺上皮细胞由内层管腔层和外层肌上皮细胞和/或基底细胞组成。腺上皮细胞由多能祖细胞发育而来,在成体组织稳态过程中逐渐被单能干细胞取代。当在没有管腔细胞的情况下从自然环境中取出时,基底干细胞表现出更大的分化潜力,产生管腔细胞,并在小鼠中产生功能性乳腺。
(e) 分化细胞恢复到干细胞样状态的能力对肿瘤发生具有重要意义,一些致癌驱动因素会影响肿瘤发生过程中的可塑性。肿瘤抑制因子(如TP53, RB1和PTEN)可调节发育分化程序,当其失调时,与癌症有关。在腺上皮中,单能性的基底和管腔干细胞在肿瘤起始时可以重新获得多能性。在小鼠前列腺肿瘤发生过程中,基底细胞中的Pten缺失促进了基底向管腔的转分化。
(f) 在小鼠乳腺中, 管腔祖细胞中BRCA1的失活可导致基底样乳腺癌,表现出基底和管腔标记物的异质性表达。致癌性的PIK3CH1047R的表达在肿瘤发生早期可诱导乳腺谱系限制性祖细胞的多能性,为肿瘤内异质性奠定了基础。
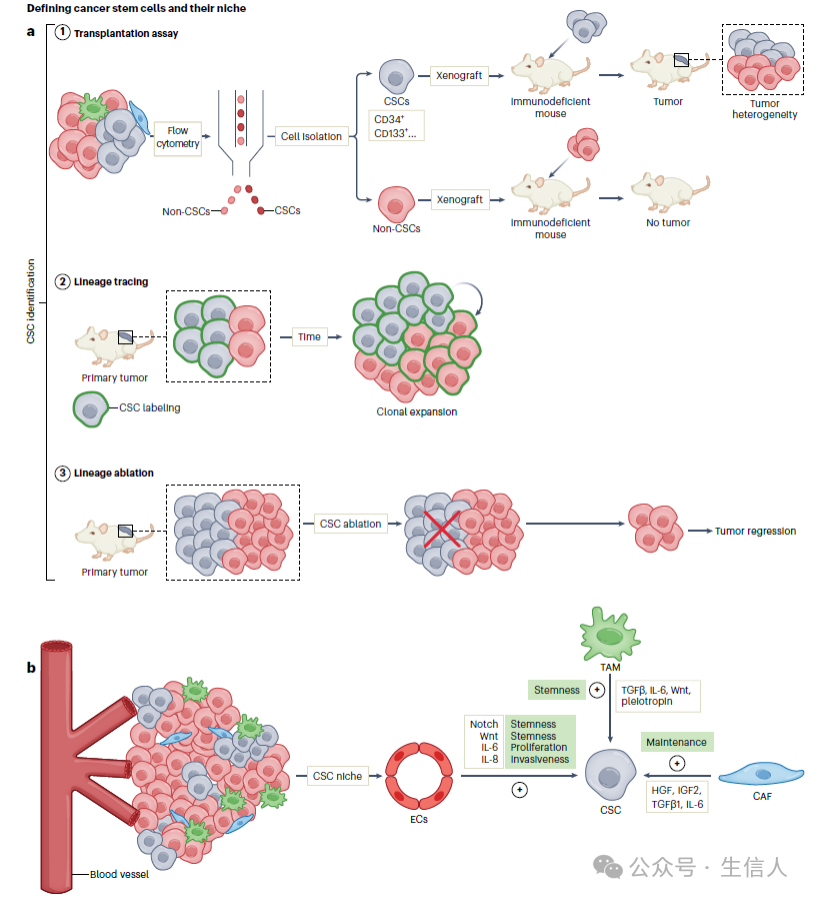 图2 肿瘤干细胞(CSC)的定义及其生态位
图2 肿瘤干细胞(CSC)的定义及其生态位 (a) 鉴定CSCs的功能策略包括三个:
① 移植试验(将通过荧光激活细胞分选,FACS)分离的肿瘤亚群移植到免疫缺陷小鼠中。如果移植了CSCs,就会形成肿瘤,并且会再现肿瘤的异质性,而非CSCs在移植后形成肿瘤的效率较低。
② CSCs的谱系追踪。
③ 谱系消除。如果CSCs被消除,剩余的亚群将无法维持肿瘤生长,肿瘤将萎缩。
(b) CSCs与其微环境之间的对话对维持肿瘤生长至关重要。CSCs 由肿瘤相关的成纤维细胞(CAFs)、内皮细胞和免疫细胞组成的生态位支持,这些生态位从外在上促进肿瘤的干性。
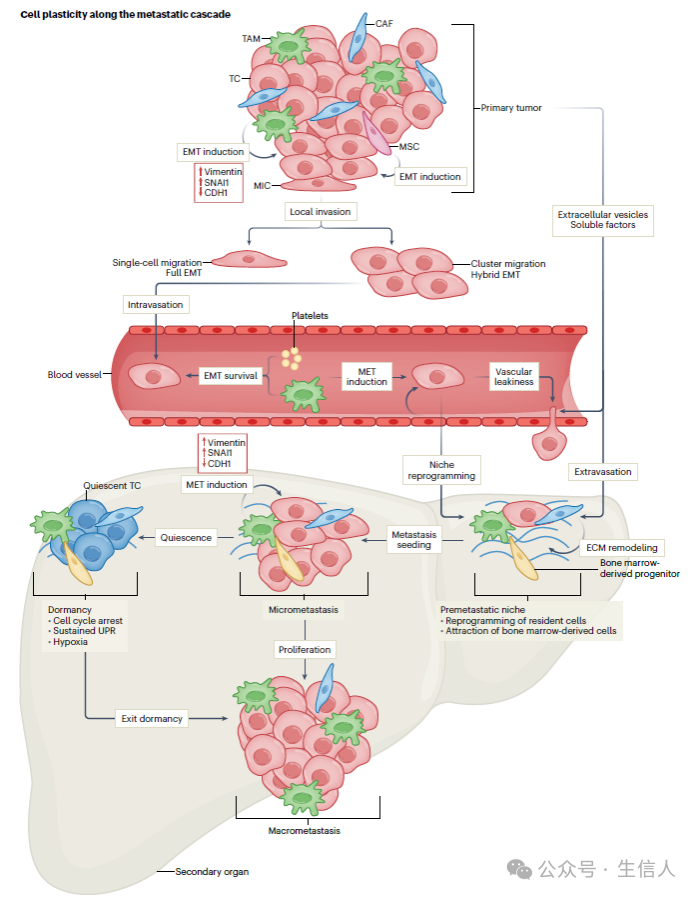 图3 沿转移级联反应的细胞可塑性
图3 沿转移级联反应的细胞可塑性 转移的发生是一个多步骤的级联过程,包括癌细胞从原发肿瘤脱离、局部浸润到周围组织、内渗到血液或淋巴管、外渗、在继发器官定植和继发肿瘤生长。
越来越多的证据表明,只有某些肿瘤细胞亚群,即转移起始细胞(MIC),能够形成转移。MIC具有高度可塑性,在整个转移级联中显示出不同程度的干性、EMT和代谢可塑性。
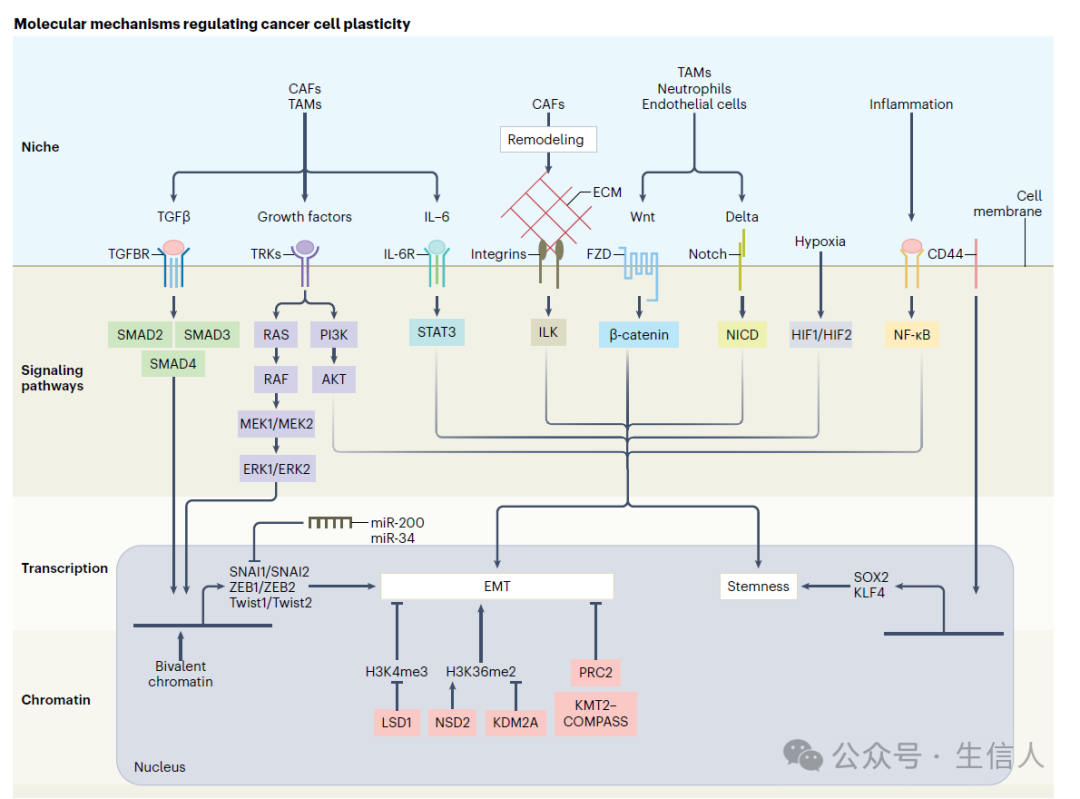 图4调节癌细胞可塑性的分子机制
图4调节癌细胞可塑性的分子机制
癌细胞的可塑性在细胞外受到来自微环境的信号调控,在细胞内则通过信号通路、转录程序和染色质重塑进行调控。
TGFβ和RAS-MAPK通路可以共同作用诱导EMT。CD44和Wnt调节干性,而Notch、JAK-STAT和整合素以依赖周围环境的方式作用于干性和EMT。缺氧诱导干性,而NF-κB通过其在炎症中的作用参与可塑性。
上述这些通路激活了EMT中涉及的关键转录因子(如SNAI1、SNAI2、ZEB1、ZEB2、Twist1、Twist2)和干性涉及的关键转录因子(如SOX2、KLF4)调控的转录程序。
上述这些关键转录因子的作用可以通过microRNA(如ZEB-miR-200和SNAI1-miR-34)的负反馈回路来调节。LSD1可以去除具有转录活性的H3K4me3组蛋白标记,并与SNAI1协同沉默上皮基因。NSD2和KDM2A分别作为在EMT期间增加的组蛋白标记H3K36me2的写入器和擦除器,从而表现出拮抗作用。PRC2和KMT2-COMPASS对调节上皮状态至关重要。
 图5抗癌治疗中基因诱导的耐药和非基因耐药
图5抗癌治疗中基因诱导的耐药和非基因耐药
预先存在的 (a)或获得的 (b)突变可以赋予内在的遗传耐药性,通过这种耐药性,突变的肿瘤细胞可以在特定的治疗方案下表现出克隆选择、存活和增殖。
(c) 尽管治疗耐药性被认为完全是肿瘤细胞基因改变的结果,但越来越多的证据表明,在没有突变的情况下仍然存在药物的耐受性。非遗传药物耐受可以通过转录选择在治疗过程中获得耐药持久性(drug-tolerant persistent, DTP)休眠状态的启动细胞而发生,并可能导致治疗后肿瘤复发。
(d) 非遗传性药物耐受可以通过适应治疗压力而发生,通过这种压力,可塑性肿瘤细胞在治疗后获得DTP静止状态,并可能导致治疗后肿瘤复发。
(e) 靶向DTP状态下激活的信号通路能够根除DTP。黑色素瘤中用BRAF和MEK抑制剂(BRAFi,MEKi)治疗诱导的DTP状态依赖于FAK信号传导,这种状态的转录程序在很大程度上是由核受体(类视黄醇X受体,RXR)驱动的。因此,DTP状态可以通过FAK抑制和RXR拮抗(FAKi,RXRi)来靶向。然而,新发突变仍可能导致遗传耐药性和肿瘤复发。
小结一下,本文综述了细胞可塑性在肿瘤发生、发展、转移和治疗抵抗中的重要性。 不同的可塑性模式通过增殖状态和CSCs参与维持肿瘤生长。可塑性还允许肿瘤细胞逃避选择压力并克服治疗。更好地了解调节可塑性的肿瘤细胞内在和外在机制,可以为新的治疗策略开辟道路,并在不久的将来提高患者的生存率。
今天分享的第二篇文章于 2024年 发表在 Cancer and Metastasis Reviews (影响因子=7.7002),题目为“ Multiomics approach towards characterization of tumor cell plasticity and its significance in precision and personalized medicine ”。这篇文章阐述了细胞可塑性在产生肿瘤内异质性中的作用,并重点阐述了使用多组学方法表征癌细胞可塑性特性的可能性和益处。文章还指出,有必要整合不同细胞组织水平(DNA、RNA、蛋白质、代谢物、表观遗传学等)的碎片证据,以促进对不同形式的可塑性和细胞类型的表征。目前对肿瘤异质性机制的理解尚未达成一致,了解这些机制对于寻找早期发现转移的靶点和有效的治疗干预是必要的。随着精准和个性化医疗的进步,这些努力可能会转化为肿瘤患者尤其是处于转移期的肿瘤患者更好的临床结局。

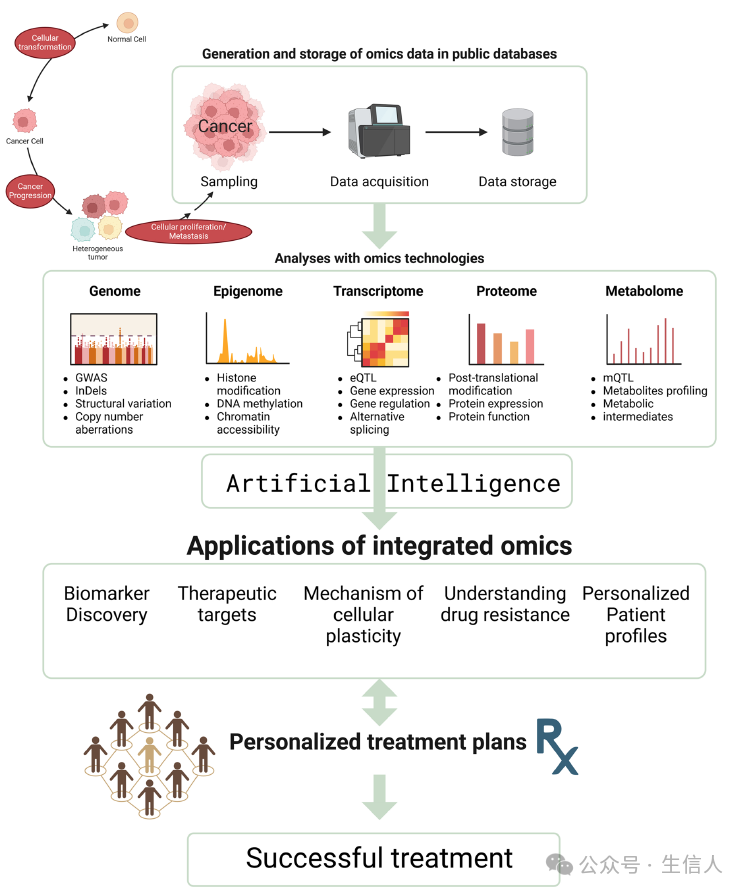 图1 多组学用于表征细胞可塑性以帮助精确和个性化的癌症治疗
图1 多组学用于表征细胞可塑性以帮助精确和个性化的癌症治疗
链接:http://www.lewenyixue.com/2024/11/28/%E8%BF%90%E7%94%A8%E5%A4%9A%E7%BB%84%E5%AD%A6%E6%96%B9%E6%B3%95%E8%A1%A8%E5%BE%81%E8%82%BF%E7%98%A4%E7%BB%86%E8%83%9E%E7%9A%84%E5%8F%AF%E5%A1%91%E6%80%A7/