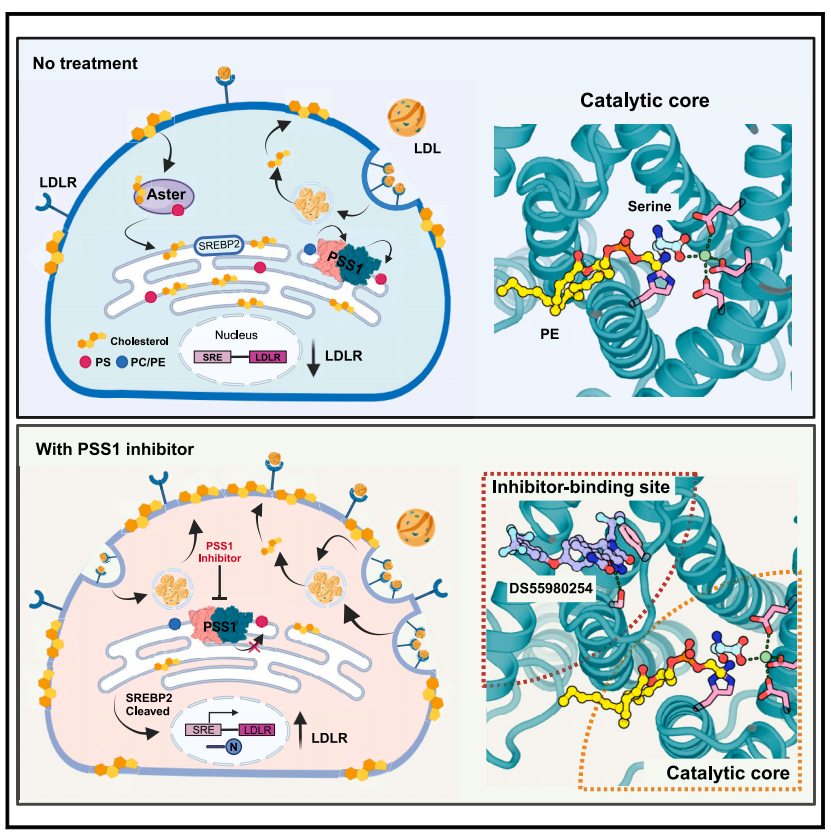
在哺乳动物细胞中,两种磷脂酰丝氨酸合成酶驱动磷脂酰丝氨酸的合成。 Ptdss1基因的功能获得突变导致PS产生增加,导致Lenz-Majewski综合征(LMS)。最近,药理抑制PSS1已被证明可以抑制肿瘤的发生。
2024年8月28日,德州大学李晓淳团队 在 Cell 在线发表题为 “ Molecular insights into human phosphatidylserine synthase 1 reveal its inhibition promotes LDL uptake ” 的研究论文, 该研究 报道了野生型人类PSS1 (PSS1 WT )、引起LMS的Pro269Ser突变体(PSS1 P269S )和PSS1 WT 与其抑制剂DS55980254复合物的冷冻电镜结构。
PSS1含有10个跨膜螺旋(TMs),其中TMs 4-8在管腔小叶中形成催化核心。这些结构揭示了PSS1的工作机制类似于膜结合O-酰基转移酶家族的假设机制。此外,PS和DS55980254都可以变变抑制PSS1,并且DS55980254的抑制激活了SREBP通路,从而增强了LDL受体的表达,增加了细胞对LDL的摄取。 这项工作揭示了哺乳动物PS合成的机制,并表明选择性PSS1抑制剂具有降低血液胆固醇水平的潜力。
另外,2024年5月23日, 德克萨斯大学西南医学中心分子李晓淳团队在 Nature Communications 在线发表题为“ Structure and inhibition of the human lysosomal transporter Sialin ”的研究论文, 该研究 展示了人Sialin的冷冻电镜结构 :载脂蛋白细胞质开放,载脂蛋白管腔开放, NAAG 结合和抑制剂结合。 结构表明,带正电荷的细胞质开放前庭可容纳NAAG或 Sialin 抑制剂Fmoc-Leu-OH,而其腔体可能结合唾液酸。 此外,功能分析和分子动力学模拟鉴定了结合唾液酸和NAAG的关键残基。 因此,该研究揭示了NAAG和唾液酸运输的基本构象状态,展示了SLC17转运体的工作模型。
2023年10月17日, 德克萨斯大学西南医学中心分子李晓淳及 齐晓峰 共同通讯在 Cell 在线发表题为“ Molecular basis of Wnt biogenesis, secretion, and Wnt7-specific signaling ”的研究论文,该研究 通过对参与中枢神经系统血管生成和血脑屏障维持的Wnt7a的结构和功能分析,阐明了Wnt7a的生物发生原理和Wnt7特异性信号传导。 Wnt7a-WLS复合物与钙网蛋白(CALR)结合,揭示了在Wnt生物发生过程中,CALR作为伴侣促进Wnt从PORCN转移到WLS。结构、功能分析和分子动力学模拟表明,Wnt结合WLS的核心磷脂调节Wnt和WLS之间的关联和解离,提示脂质介导的Wnt分泌机制。 最后,Wnt7a与细胞表面Wnt7共受体RECK结合的结构揭示了RECKCC4如何参与Wnt7a的N端结构域来激活Wnt7特异性信号传导。
2023年9月28日,德州大学西南医学中心李晓淳及Eric Olson共同通讯在 Nature Structural & Molecular Biology 上发表了题为“ Cryo-EM structures of Myomaker reveal a molecular basis for myoblast fusion ”的研究论文,该研究 报道小鼠Myomaker (mMymk)和Ciona robusta Myomaker (cMymk)的冷冻电镜结构。Myomaker含有7个跨膜螺旋(TMs),采用G蛋白偶联受体样折叠。TMs 2-4形成二聚体界面,而TMs 3和5-7创建一个脂质结合位点,保持磷脂的极性头部,并允许烷基尾部插入Myomaker。cMymk和mMymk的相似性表明myomaker介导的细胞融合机制在进化上遥远的物种中是保守的。功能分析证明了二聚体界面和脂质结合位点对融合活性的重要性,异源细胞-细胞融合实验显示了Myomaker原蛋白的跨细胞相互作用对成肌细胞融合的重要性。 总之,该研究的发现为成肌细胞融合的过程提供了结构和功能上的见解。
2023年5月23日,美国圣裘德儿童研究医院李佳学及德克萨斯大学西南医学研究中心李晓淳共同通讯在 Cell 上发表了题为“ Structural and Functional insight into Spns2-mediated transport of sphingosine-1-phosphate ”的文章, 该研究报道了淋巴管/血管内皮细胞中由 Spns2介导的磷酸鞘氨醇的转运机制。
2022年9月15日,斯坦福大学医学院冯亮、加州大学圣克鲁斯分校Glenn Millhauser及德克萨斯西南医学研究中心李晓淳共同通讯在 Cell 上发表了题为“ Structure and mechanism of human cystine exporter cystinosin ”的文章, 该研究报道了溶酶体胱氨酸的转运机制。
2022年7月13日,德克萨斯大学西南医学中心分子李晓淳团队在 Nature 在线发表题为“ Mechanisms and inhibition of Porcupine-mediated Wnt acylation ”的研究论文,该研究报告了人类 PORCN 的四种冷冻电子显微镜结构:与棕榈油酰辅酶 A (palmitoleoyl-CoA) 底物的复合物;与目前处于临床试验中的抗癌药物 PORCN 抑制剂 LGK974 的复合物;具有 LGK974 和 WNT3A 发夹 2 (WNT3Ap) 的复合物;以及与合成的棕榈油酰化 WNT3Ap 类似物的复合物。 这些结构表明, 在所有 Wnt 配体中都非常保守的 WNT3A 的发夹 2 从管腔侧插入 PORCN,而棕榈油酰辅酶 A 从胞质侧进入酶。 催化组氨酸触发不饱和棕榈油酰基转移到 Wnt 发夹 2 上的目标丝氨酸,这得益于两个底物的接近。抑制剂结合结构表明 LGK974 占据了棕榈油酰辅酶 A 结合位点以阻止反应。因此, 这项工作为 Wnt 酰化提供了一种机制,并推动了用于癌症治疗的 PORCN 抑制剂的开发。






赶快来坐沙发