Cancer Cell最新万字综述:肿瘤免疫逃逸的标志
尽管在癌症早期,恶性细胞可以被免疫系统有效地消除,但也可以通过遗传或表观遗传改变,实现明显的免疫逃逸。今天,小编要和大家分享一篇 2024年10月 发表在 Cancer Cell(IF:48.8) 上的文章,作者根据肿瘤免疫的“3E”模型提出了新的“3C”模型来回顾癌症免疫逃避的特征、与这些特征相关的宿主改变以及预防或逆转此类免疫逃避机制以达到治疗目的的有前景的策略。

“3C”模型
经典的免疫逃逸的“3E”模型,即宿主免疫系统 消除(eliminates) 恶性细胞前体,并在动态 平衡(equilibrium) 中包含微观肿瘤,防止癌症生长,直到肿瘤细胞获得能够 逃避(escape) 免疫的遗传或表观遗传改变。这种免疫逃避表型源于多种机制,这些机制可归类为新的 “三C” 概念框架:
(1) 伪装(camouflage) ,隐藏癌细胞免遭免疫识别;
(2) 强制(coercion) ,直接或间接干扰免疫效应细胞,
(3) 细胞保护(cytoprotection) ,保护恶性细胞免受免疫细胞毒性。
 图1. 肿瘤免疫逃逸的“3C”模型
图1. 肿瘤免疫逃逸的“3C”模型 一、伪装(camouflage)
恶性细胞逃避免疫监视的一种主要策略依赖于它们躲避免疫效应细胞的能力,即避免被免疫效应细胞定位或识别。这种伪装可能是由于 抗原加工和呈递的缺陷、与免疫原性细胞死亡(ICD)相关或无关的趋化因子的有限分泌、 或 防止免疫浸润的基质屏障的产生 而导致的。
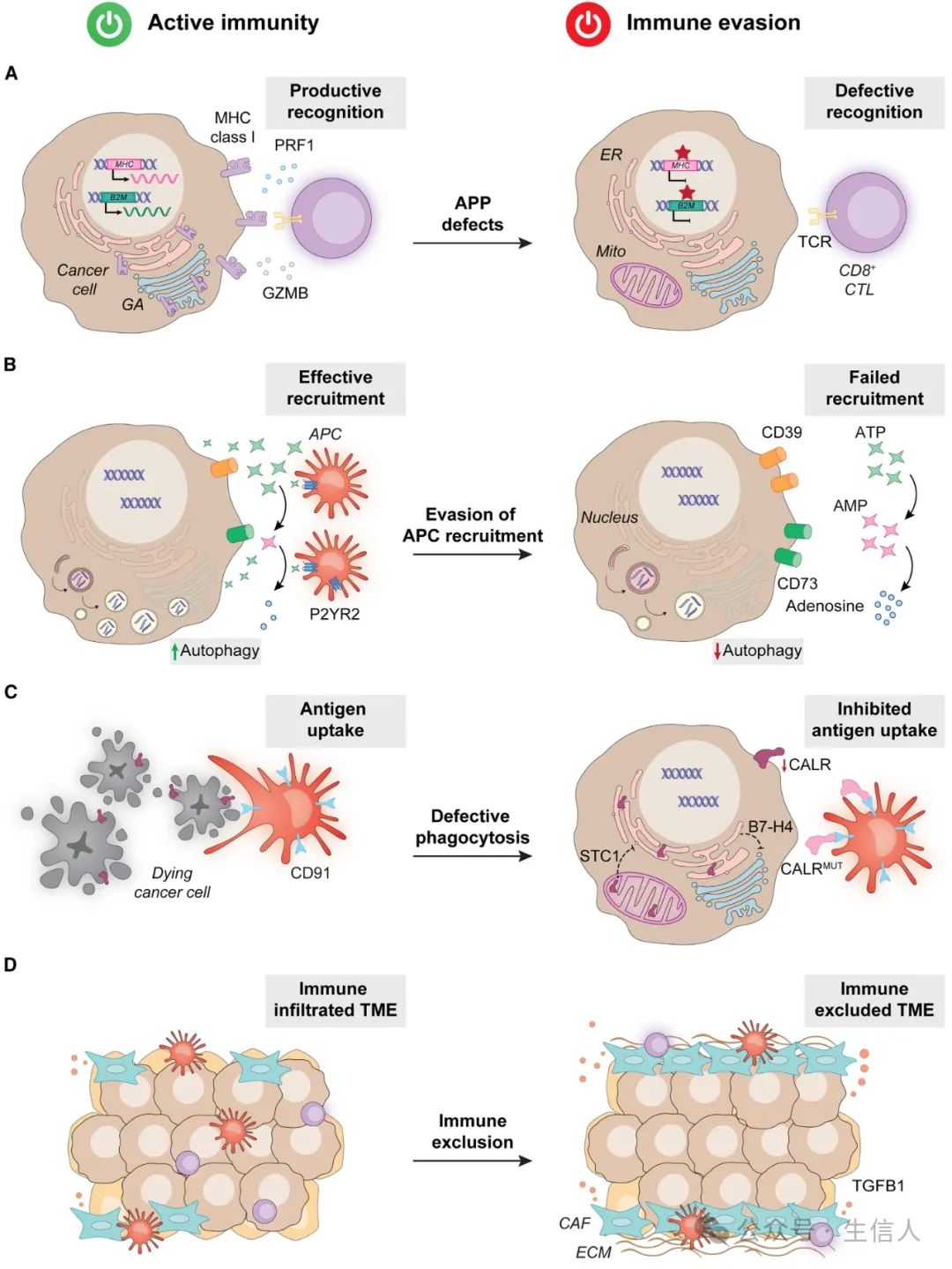 图 2 . 癌症免疫逃避中的伪装
图 2 . 癌症免疫逃避中的伪装 1.抗原加工和呈递缺陷
与正常细胞相比,突变和非突变事件会导致细胞的抗原发生显著变化,然而,恶性细胞在细胞表面正确处理和暴露此类新抗原的分子机制通常存在缺陷,从而使它们能够避免免疫识别。
首先,肿瘤细胞可以通过遗传机制获得抗原加工和呈递(APP)机制的缺陷。 例如,编码MHC分子或专性MHC I类相互作用蛋白β-2微球蛋白(B2M)的基因突变,以及包含MHC位点的6p染色体杂合性丢失(LOH)。
其次,恶性细胞中的抗原呈递缺陷也可由表观遗传机制引起。 例如抑制性组蛋白乙酰化标记的积累以及随之而来的APP成分的下调,包括蛋白酶体20S亚基LMP7、LMP2、与抗原加工相关的转运蛋白TAP1和TAP2,以及通过在MHC 编码基因的启动子区域建立由EZH2和甲基转移酶SETDB1催化的抑制性H3K9me3和H3K27me3标志。值得注意的是,免疫逃避性恶性肿瘤通常表现出特定基因组区域DNA甲基化减少,从而促进部分甲基化结构域(PMD)的形成。这种PMD与多种免疫调节基因的沉默有关,包括APP相关基因。
再者,翻译后修饰也会导致癌细胞中的APP缺陷。 例如,自噬受体NBR1可通过自噬引起MHC I类分子的降解,蛋白酶PCSK9通过不依赖于胆固醇的机制促进MHC I类分子的溶酶体降解,SUSD6-TMEM127-WWP2的泛素-蛋白酶体系统降解MHC I类分子。
最后,癌细胞还可下调驱动免疫识别的显性新抗原的遗传或表观遗传。 例如,新抗原表达基因在启动子高甲基化时被沉默,而表观遗传修饰剂如阿扎胞苷和地西他滨可部分恢复新抗原表达和免疫疾病控制。
2.逃避APC招募
恶性肿瘤在各种治疗的压力下发生细胞死亡,而这些细胞死亡会引发与效应器阶段和免疫记忆(即ICD)相关的抗原特异性免疫应答。ICD 依赖于应激反应途径的死前激活,包括(但不限于)自噬和综合应激反应 (ISR),最终为抗原呈递细胞(APC)或其前体产生趋化、促吞噬和免疫刺激信号,这些信号统称为损伤相关分子模式(DAMP)。 由于化学趋化缺陷或促吞噬信号暴露导致癌细胞和APC 之间缺乏相互作用 ,这也是一种常见的伪装机制。
化学趋化剂的释放缺陷
接受ICD的恶性肿瘤细胞释放ATP和ANXA1,它们在与嘌呤受体P2RY2和甲酰肽受体FPR1结合后分别作为APC的中程和短程趋化剂。癌细胞通过多种机制来抑制ICD相关的ATP和ANXA1释放。此外,ICD还与将免疫效应细胞募集到TME的趋化因子的释放相关,特别是CXCL10和CCL2。另外,在某些肿瘤中,与ICD无关的化学趋化剂的释放也可能存在缺陷,例如CCL4和CCL20的转录抑制和分泌缺陷。
ICD驱动的吞噬作用缺陷
ICD相关的CALR在癌细胞表面的暴露,会影响ISR和eIF2α磷酸化,在与LDL受体相关蛋白LRP1结合后向APC提供有效的促吞噬信号。
癌细胞通过以下方式避免CALR介导的APC吞噬作用:
(1)下调CALR表达;
(2) 在STC1过度表达时在线粒体捕获CALR;
(3)上调含有T细胞激活抑制剂VTCN1,从而抑制 eIF2α磷酸化;
(4)分泌截短形式的CALR,竞争性抑制表面暴露的CALR和LRP1之间的相互作用。
癌细胞还可通过特异性免疫和基质细胞介导的,或改变细胞外基质(ECM)来重塑TME,从而阻碍免疫效应细胞招募至TME,以此进行伪装。
TME的三个主要细胞群,癌症相关成纤维细胞(CAF)、肿瘤相关巨噬细胞(TAM)和肿瘤相关中性粒细胞(TAN)积极促进排斥,从而对基于ICI的免疫疗法产生抵抗。此外,WNT和CDK4/6信号传导也参与了免疫效应细胞招募的排斥。
伪装失败的恶性细胞(因此被发现并被识别为被免疫系统转化)可以通过 抑制免疫效应细胞的活性 来逃避抗癌免疫,这些免疫效应细胞包括但不限于DC、NK细胞、TH1极化的CD4+ T细胞和CD8+ CTL,以及通过 促进免疫抑制细胞的活性 ,例如CD4+ CD25+ FOXP3+调节性T(TReg)细胞、特定TAM亚群和MDSC。这种强制活性可能是多种变化的结果, 这些变化涉及癌细胞表面免疫调节配体表达的变化、DAMP或促炎细胞因子信号传导的缺陷和/或TME中免疫调节代谢物的释放。
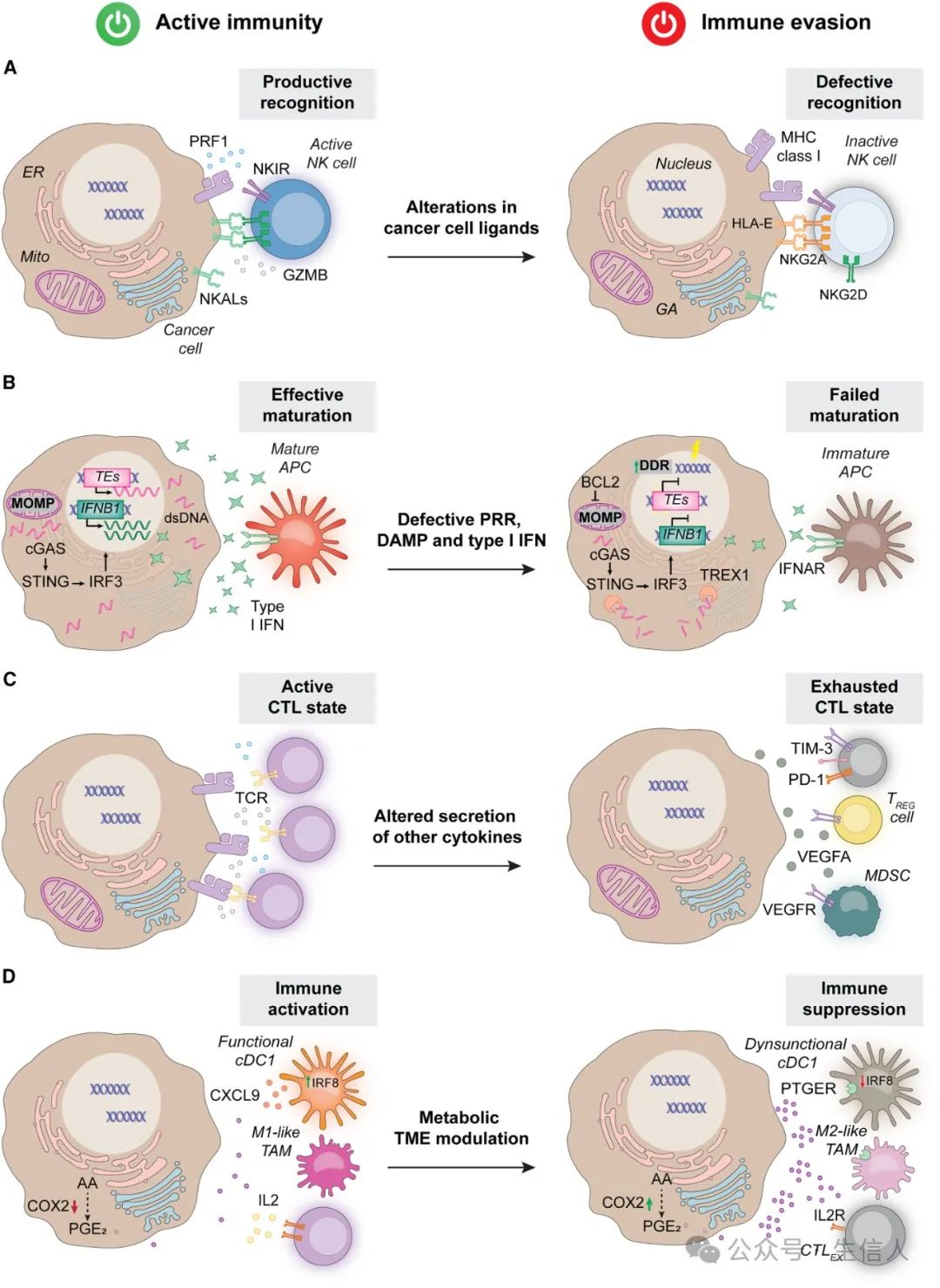 图 3 . 癌症免疫逃避中的强制
图 3 . 癌症免疫逃避中的强制 大多数肿瘤细胞的特征是表面表型改变,包括支持伪装的MHC水平降低(前面提到),以及具有免疫刺激或免疫抑制作用的免疫细胞受体配体表达的变化。
最著名的PD-L1 ,是癌细胞用来支持直接强制的最具特征性的表面分子,很大程度上反映了它抑制CD8+ CTL效应子功能的能力。PD-L1水平受启动子区域DNA 和组蛋白的抑制标记的表观调控,也受真核翻译起始因子EIF4E的翻译调控,还受JAK-STAT信号介导的的磷酸化修饰等蛋白翻译后修饰调控。
NK细胞激活配体NKAL 也被癌细胞下调以逃避NK细胞的清除,例如MHC I类多肽相关序列MICA、MICB和UL16结合蛋ULBP。此外,癌细胞还可以通过 过度表达NK细胞抑制配体NKIL 来胁迫NK细胞,例如HLA-E。
其他免疫调节配体还包括PVR细胞粘附分子CD155、CD226(也称为DNAM-1)、颗粒酶B(GZMB)、B7-H4、半乳糖凝集素LGALS9等等。
cGAS-STING1信号传导缺陷
cGAS是胞浆双链DNA(dsDNA)的传感器,在STING和TBK1信号传导后,引发由干扰素调节因子IRF3、IRF7和NF-κB执行的促炎转录程序。cGAS-STING1信号传导通过涉及蛋白质激酶LKB1缺失、甲基转移酶PRMT5和SUV39H1高表达等表观遗传调控,或涉及蛋白激酶DAPK3等泛素化的翻译后修饰机制受损后,赋予恶性细胞免疫逃避特性。
病毒拟态受损
内源性逆转录病毒(ERV)和其他转座元件(TE)也是胞质核酸的重要来源,它们通过cGAS或胞浆RNA传感器(如RNA传感器RIG-I或IFIH1)驱动I型IFN 反应但也可能产生新抗原。然而,恶性肿瘤细胞,尤其是CSC,会沉默ERV和其他TE的表达,这主要是通过DNA和组蛋白甲基转移酶实现的。此外,p53和 CDK4/6也参与了ERV的转录调节以及随后的I型IFN反应。
另外,癌细胞通过确保严格的线粒体检查点,例如过表达BCL2的内源性抑制剂和增强线粒体自噬,或者过表达一些胞质核酸酶,来降解胞质mtDNA和源自破裂微核的胞质dsDNA,以避免潜在干扰性dsDNA在胞质中的积累。
DAMP信号传导缺陷
非组蛋白染色质结合蛋白高迁移率族蛋白HMGB1是主要的ICD相关DAMP之一,其微环境积累在与Toll样受体TLR4和高级糖基化末端结合后介导免疫刺激作用。在肿瘤进展过程中,HMGB1和其他DAMP的表达缺失,或者免疫抑制性DAMP的分泌导致DAMP信号传导缺陷。
细胞凋亡信号传导
半胱天冬酶caspase激活具有免疫抑制作用,可能与干扰性mtDNA的胞质积累有关,或者抑制MOMP驱动的I型IFN分泌。
慢性I型干扰素信号传导
急性和消退性炎症过程通常促进免疫监视,而慢性和惰性炎症通常支持免疫逃避,一些肿瘤细胞启动弱且慢性的I型干扰素信号传导以支持局部免疫抑制和加速疾病进展。
肿瘤还可以通过其分泌组的改变来逃避抗癌免疫,这些改变涉及 抗炎细胞因子的累积产生 和/或 I型干扰素之外的促炎因子的释放缺陷 。抗炎细胞因子包括CCL2、CCL5、CCL8、CCL28、CXCL8、IL33和VEGFA,从而建立局部和/或全身免疫抑制;I型干扰素之外的促炎因子包括IL1B和IL10,也可以促进局部免疫抑制。
与正常细胞相比,所有癌细胞都表现出相当显著的代谢重组,以积极促进免疫抑制。 几种常见的代谢途径包括:
(1) ATP 和腺苷 :细胞外ATP水解和随后的腺苷积累通过与T细胞表面的腺苷受体ADORA2A结合,可有效限制TCR信号传导和IFNG的产生
(2) 犬尿氨酸 :由IDO1和TDO2催化的色氨酸转化为犬尿氨酸,可通过芳基碳氢化合物受体AHR依赖性机制招募M2样TAM,促进CCL5分泌,或通过旁分泌AHR信号传导抑制CD4+和CD8+ T细胞的增殖和功能。
(3) 葡萄糖和谷氨酰胺 :TME的不同细胞区室优先吸收不同的营养物质,且具有肿瘤类型依赖性,从影响能量代谢和存活,效应T细胞的激活和增殖等方面促进或抑制抗癌免疫。
(4) 乳酸 :细胞外乳酸常常通过促进TME酸化和抑制免疫效应细胞(包括 CD8+ CTL和NK细胞)的增殖和功能来促进免疫逃避。
(5) 其他TCA循环中间体 :癌细胞通过TCA循环酶的突变导致免疫抑制代谢物的积累,例如富马酸水合酶FH缺陷导致富马酸分泌增加,乙酰辅酶A 合成酶ACSS2的抑制导致微环境中醋酸盐的积累,IDH1的功能获得性突变导致D-2-羟基戊二酸(D-2HG)的积累,不仅为癌细胞提供生长优势,而且使它们具有免疫逃避能力。
(6) 酮体 :细胞在缺乏足够碳水化合物供应的情况下产生替代燃料--酮体,某些情况下,通过转录因子KLF5依赖性机制从CAFs 释放的CXCL12会导致M2样TAMs、MDSCs和TREG细胞被招募到TME,促进免疫抑制。
(7) 蛋氨酸和丝氨酸 :蛋氨酸是SAM的来源之一,SAM和其他蛋氨酸代谢副产物如 5-甲硫腺苷(MTA)的积累可通过促进T细胞耗竭来介导免疫抑制作用;而丝氨酸限制可损害线粒体检查点导致免疫抑制。
(8) 脂质 :肿瘤细胞表现出脂质代谢的重大重组,以支持加速增殖,也会促进免疫逃避,涉及FASN介导的脂质合成、脂肪酸氧化、CD36介导的脂肪酸摄取,都在某些肿瘤中会引起免疫抑制。
(9) 生物活性脂质 :包括PGE2和溶甘油磷脂,可通过降低肿瘤内cDC1或直接影响肿瘤浸润CD8+ CTL的功能和存活等机制支持免疫逃避。
(10) 维生素和视黄酸 :主要是通过影响各种免疫效应细胞的激活,如NK细胞、单核细胞、树突细胞来调节抗癌免疫。
(11) 钾 :主要是调节T细胞,尤其是CD8+ CTL的功能。
CD8+ CTL和NK细胞在建立“免疫突触”后杀死恶性细胞。
“免疫突触”是一种高度组织化的结构,将免疫效应细胞与其靶标物理连接起来,并能够极化
(1)释放细胞毒性分子,包括GZMB和PRF1;
(2)死亡受体配体的表达,例如Fas配体(FASLG);
(3)释放肿瘤靶向细胞因子,特别是IFNG。肿瘤细胞可以逃避效应细胞的免疫消除,效应细胞通常将它们识别为转化的,因此通常通过多种细胞保护机制参与细胞毒性和分泌功能,这些细胞保护机制涉及 免疫突触形成的缺陷,下游细胞死亡途径的激活,或依赖于自噬等补偿反应的启动。
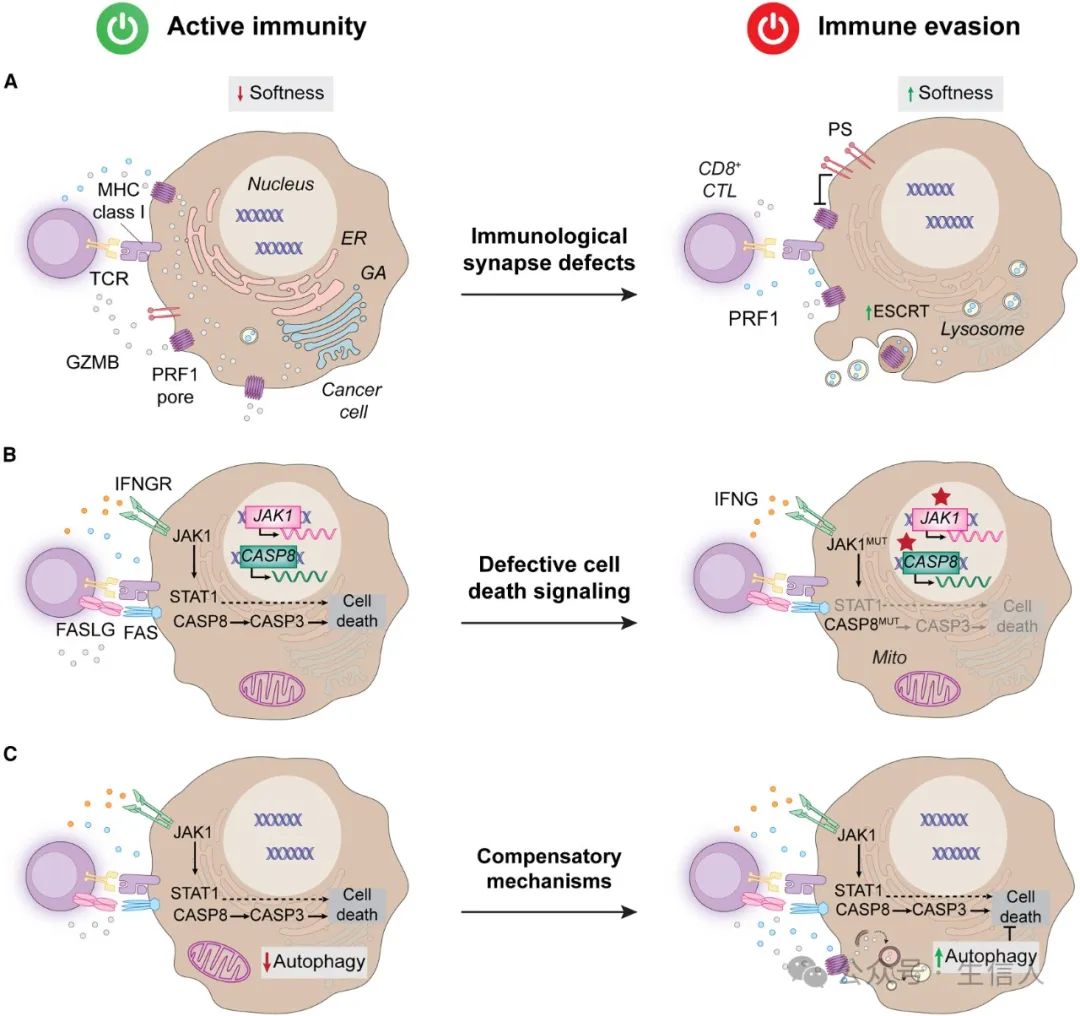 图 4 . 癌症免疫逃避中的细胞保护
图 4 . 癌症免疫逃避中的细胞保护
癌细胞通过机械特性逃避免疫细胞毒性,从而导致对GZMB和PRF1的敏感性降低。这种机械特性包括TCR的柔软性导致免疫突触处PRF1孔形成的收缩力不足、广泛的肌动蛋白重塑导致GZMB进入细胞质的机会减少、肌动蛋白细胞骨架收缩不足阻止免疫突触的快速溶解,导致效应细胞保留和其他靶标的参与受到限制、利用免疫突触处转运所需的内体分选复合物ESCR修复PRF1孔,从而限制GZMB的进入等。
恶性细胞已被证明经常通过表观遗传机制下调FAS,从而产生对NK细胞毒性的抵抗力。此外,完整的TNF和IFNG信号传导缺陷与免疫治疗耐药性相关,涉及凋亡caspase的激活、JAK-STAT信号传导的激活、细胞周期的调控、铁死亡脂质氧化,以及细胞焦亡等多种复杂机制。
肿瘤细胞还可以激活许多细胞范围的通路,使它们抵抗TNF、IFNG和PRF1-GZMB系统的杀伤,例如激活自噬,通过激活STAT磷酸化从而使溶酶体GZMB降解。
TFGB1还可以通过涉及SOX4的机制限制癌细胞对CD8+ CTL的敏感性,从而促进细胞保护,可能是通过降低对IFNG的敏感性。此外,对免疫细胞毒性的累积抵抗力也与获得静止表型有关,其特征是转化细胞低氧反应的转录特征。
免疫疗法已经进入常规临床应用 ,包括:
(1)一些以PD-1、PD-L1、CTLA4或 LAG3为靶点的ICIs,它们通过抑制由癌细胞表达的共抑制配体驱动的胁迫而发挥作用;
(2)CAR T细胞和双特异性抗体,它们可以规避伪装;
(3)一些重组细胞因子和 TLR激动剂,它们通过针对细胞因子和DAMP信号转导中免疫抑制转变驱动的胁迫而发挥作用。
此外,还有 一些抗癌疗法以免疫不可知论方式被发现(通过多种机制)抑制了免疫逃避 ,包括:
(1)一些细胞毒药物,如蒽环类和奥沙利铂;
(2) 一些靶向抗癌药物,包括各种酪氨酸激酶抑制剂(TKIs)、肿瘤靶向抗体、表观遗传修饰剂和BCL2抑制剂venetoclax;
(3)至少在某些情况下的病灶放疗。
除了已建立的方法之外,许多临床试验现已开放,以 研究抑制免疫逃避的实验策略 ,其通常 旨在
(1)抑制共抑制配体的强制;
(2)抑制代谢胁迫并抑制细胞因子信号传导;
或(3)同时瞄准多个“C”。
主要是关注阻断除PD-1、CTLA4和LAG3之外的共抑制受体的强制作用,特别是TIM-3、TIGIT、VISTA和NKG2A,通常与FDA批准的PD-1或PD- L1抑制剂联合使用。
关注以癌细胞代谢重塑驱动的强制为目标的药物包括谷氨酰胺拮抗剂DRP-104、MCT1抑制剂AZD3965、SAM给药、FASN抑制剂TVB-2640。
通过DDR抑制剂和模式识别受体(PRR)激动剂增强免疫刺激细胞因子信号传导的药物主要包括ATR和ATM信号转导器抑制剂和STING激活剂。
FDA 批准的表观遗传修饰剂等多种药物,这些药物同时针对伪装和强制,如氮胞苷、地西他滨和多种 HDAC 抑制剂。然而,需要注意的是, 许多免疫细胞的生存、增殖和效应功能也依赖于表观遗传机制,这些表观遗传药理学修饰剂也可能产生不必要的免疫抑制作用 ,开发精确的靶向策略就显得尤为重要了。
简而言之,本综述提出了一个新的概念框架来对癌症免疫逃避进行分类,表明恶性细胞逃避免疫识别和消除的大多数机制涉及“三个C”:伪装、强制或细胞保护。癌细胞表现出遗传或表观遗传缺陷,同时涉及多个“C”,以实现免疫逃避和加速疾病进展。新的临床相关的策略急需开发以规避它们。
链接:http://www.lewenyixue.com/2024/10/30/Cancer%20Cell%E6%9C%80%E6%96%B0%E4%B8%87%E5%AD%97%E7%BB%BC%E8%BF%B0%EF%BC%9A%E8%82%BF%E7%98%A4/