ADC药物安全性管理——相关毒性概述
前言
抗体药物偶联物(ADC)作为一种创新的癌症治疗策略,结合了单克隆抗体的靶向性和小分子化疗药物的强大细胞毒性,旨在提高癌症治疗的精准性和效果。然而,ADC的复杂结构和多重成分使其在临床应用中面临独特的挑战和风险。
本文将详细探讨ADC相关毒性的发生机制,重点分析靶向依赖性和非靶向依赖性毒性,阐述药代动力学特征,以及影响毒性的多种因素,包括肝功能障碍、靶抗原特性、载荷类型和连接子稳定性等。通过全面了解这些毒性机制和影响因素,临床医生可以更好地评估ADC的安全性和有效性,从而优化癌症治疗方案,提高患者的疗效和生活质量。
ADC相关毒性的发生机制
与ADC相关的毒性可以分为两类:靶向依赖性和非靶向依赖性。靶向依赖性毒性由ADC在正常细胞中表达靶抗原的结合和内化引起,而非靶向依赖性毒性则由其他因素介导,包括ADC的不稳定性、非特异性摄取进入正常细胞以及抗体对正常细胞的非特异性结合 [1] 。非特异性ADC摄取通过抗原非依赖性吞饮、ADC代谢物的摄取或Fc受体介导的摄取发生,并可能导致正常细胞的非靶向毒性。
抗体药物偶联物在体内以循环成分的动态混合物形式存在,包括偶联抗体、裸抗体和游离载荷分子,从而使每种药物的临床特性复杂化。
表1. ADC及其组分的药代动力学特征
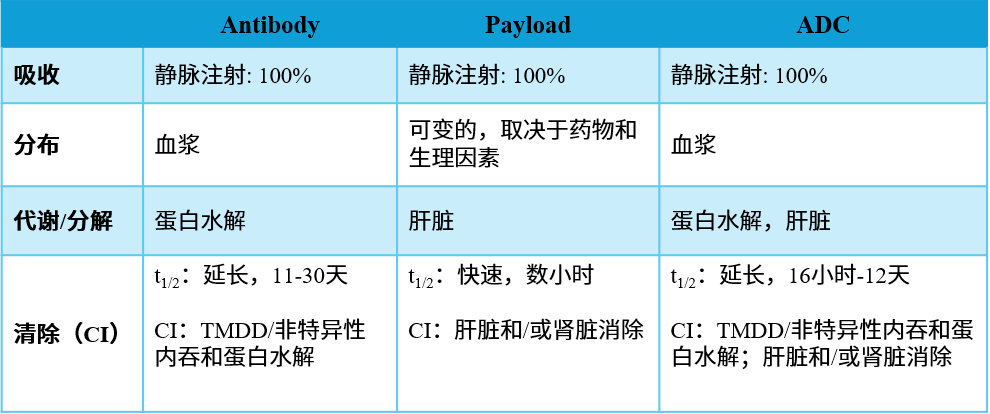
ADC的毒性可以通过上述任何一种成分介导——在正常细胞上低水平表达靶抗原、早期断裂的连接子或通过Fc受体结合介导的抗体依赖性细胞的细胞毒性 [2] 。在各种可能性中,剂量限制性毒性(DLT)和潜在药物相互作用主要与靶抗原或抗体无关,而是由载荷驱动。具有相同类型有效载荷的ADC通常具有高度相似的毒性特征和剂量限制性毒性。
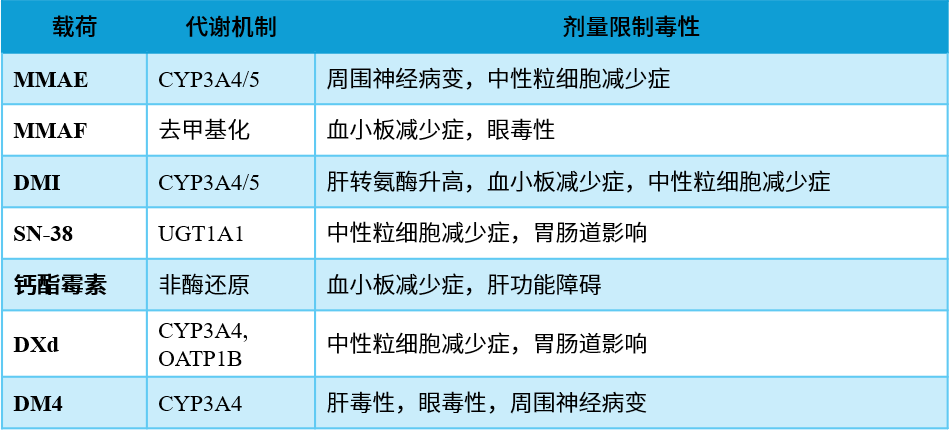
表2. 常见ADC载荷 [1]
影响ADC毒性的因素
肝脏和肾脏功能障碍对ADC及其药代动力学的影响仍是一个新兴的研究领域。现有数据显示,肝功能不全可能导致ADC和载荷的不同暴露,从而影响安全性和有效性 [3] 。
一项I期研究评估了肝功能不全患者的药代动力学,结果显示,由于清除率增加,ADC暴露减少,而MMAE暴露增加了2.3倍。然而,这种暴露差异并未与更多不良反应相关联 [4] 。另一项类似的研究显示,轻度和中度肝功能不全患者的T-DM1清除率分别短暂增加了1.8倍和4倍。重复给药后,这一情况在第3周期时恢复正常,并且没有观察到DM-1载荷浓度显著升高 [5] 。
肝功能不全导致暴露减少的总体趋势尚不完全清楚;目前提出合理的机制包括影响FcRn结合、靶点介导的药物处置(TMDD)和FcγR结合 [6] 。尽管目前尚未完全阐明确切的机制或药代动力学差异,但如果ADC的载荷在肝脏中大量代谢,临床医生应考虑在轻度到中度肝功能不全患者中调整剂量 [7] 。
理想的ADC靶抗原应高度选择性地针对肿瘤组织,而不是正常组织;然而,几乎不可能找到一个在正常组织中不表达的靶抗原。在考虑相关的靶向毒性时,肿瘤抗原靶向是关键因素。
Enfortumab vedotin是一种由针对Nectin-4的人源化IgG1抗体通过蛋白酶Val-Cit二肽连接子偶联到MMAE的ADC。其载荷和连接子相关的非靶向毒性,包括通过非特异性摄取和旁观者效应引起的中性粒细胞减少和外周神经病变。然而,载荷和连接子均不能解释独特的皮肤毒性,这些毒性包括皮肤干燥、瘙痒、水疱性皮炎以及剥脱性皮炎等。这表明靶抗原是皮肤毒性的罪魁祸首 [8] 。
例如,Nectin-4在人体皮肤角质形成细胞和皮肤附属器(包括汗腺和毛囊)中显示出均匀的弱至中等染色,从而使皮肤毒性成为可预测的靶向毒性。
外周神经病变是与微管抑制药物(如紫杉烷、长春碱、铂类化合物以及硼替佐米和含MMAE载荷的ADC)主要相关的不良反应 [9] 。此外,含MMAE载荷的ADC还表现出中性粒细胞减少的剂量限制性毒性。
基于生理学的药代动力学模型表明,MMAE从血浆中迅速消除,类似于小分子药物,但在组织、血细胞和肿瘤中分布广泛且持久,可能导致外周神经病变的高发病率 [10] 。然而,外周神经病变和中性粒细胞减少的发生概率随着ADC暴露量的增加而增加,但随着MMAE水平的增加保持平稳或减少 [11] 。临床上,外周神经病变的中位发病时间为12.4周。这表明 外周神经病变和中性粒细胞减少是MMAE通过其非特异性摄取和与抗体偶联后的长半衰期所致 [12] 。
将抗体与小分子有效载荷连接的连接子具有双重重要性,在确保ADC安全有效传递到靶细胞时提供稳定性,并在内化后可靠释放以发挥其细胞毒作用 [13] 。
Gemtuzumab ozogamicin被认为是第一代ADC,因为它使用了腙键连接子,该连接子利用N-羟基琥珀酰亚胺将calicheamicin与其抗CD33单抗结合 。酸可裂解连接子如水杨醛连接子设计为在血液中生理pH下保持稳定,但在溶酶体的酸性环境中发生水解并释放其有效载荷 [14] 。然而,这些酸不稳定的连接子在循环中发生早期释放,导致ADC聚集、半衰期缩短以及脱靶毒性增加 [15] 。
与腙键连接子相比,开发不可裂解连接子和酶可裂解连接子可以提高ADC在血清中的稳定性,减少脱靶毒性。
Ado-trastuzumab emtansine(T-DM1)是首个使用不可裂解连接子的FDA批准的ADC。临床体内研究表明,这种类型的连接子与先前ADC(如gemtuzumab ozogamicin)使用的可裂解连接子相比,毒性较小,从而减少了释放到循环中的自由DM1的数量 [16] 。一旦T-DM1与HER2的胞外结构域结合,该复合物被内化到内质网中,抗体被蛋白酶降解。有效载荷与一个带电的赖氨酸和连接子一起释放到细胞质中。由于这种带电的分子无法穿过细胞膜,因此它无法像之前描述的那样发挥旁观者效应,从而限制了非特异性毒性的可能性 [17] 。
酶可裂解连接子通过溶酶体蛋白酶降解,以确保在血清中稳定,并在内化到靶细胞后可靠释放。一项研究比较了用酸不稳定水杨醛连接子或蛋白酶敏感二肽连接子连接的mAb-MMAE偶联物的特性,发现后者与相应的水杨醛偶联物相比,在体外具有更大的特异性和较低的体内毒性 [18] 。
Brentuximab vedotin是一种针对CD30 + 的人嵌合抗体,通过蛋白酶可裂解连接子与MMAE共价连接。在与CD30 + 细胞结合和内化后,溶酶体蛋白酶从二肽连接子中释放MMAE,使其与微管蛋白结合并阻止其聚合。除了传递到其主要目标外,一小部分MMAE从抗原阳性肿瘤细胞扩散,通过旁观者效应对肿瘤微环境中的周围细胞产生细胞毒作用 [19] 。这在霍奇金淋巴瘤中尤为重要,因为微环境被认为在维持恶性霍奇金淋巴瘤细胞的生存中起着作用。
总体而言,brentuximab vedotin中的Val-Cit二肽连接子不仅通过防止MMAE有效载荷的过早释放来确保足够的安全性,还通过在CD30 + 细胞中靶向结合和释放MMAE以及未结合的MMAE向周围基质迁移来驱动霍奇金淋巴瘤的疗效。
连接子-有效载荷复合物通过化学方式偶联到抗体表面的氨基酸残基上。根据所选氨基酸残基和偶联方法的不同,DAR会有所不同。DAR可以影响ADC的安全性和有效性。尽管高DAR提高了ADC的效力,但也增加了聚集风险、清除率和循环中有效载荷的过早释放风险 [20] 。相比之下,低或广泛分布的DAR可能导致ADC的有效性降低。开发ADC的目标是确定足够的DAR并控制其分布,以优化ADC的效力和安全性。
Gemtuzumab ozogamicin通过赖氨酸偶联合成,平均DAR为2-3 [21] ;然而,约50%的抗体载有4-6个calicheamicin分子,而其余50%的抗体未与calicheamicin载荷偶联。赖氨酸残基在ADC中用于偶联载荷,因为它们丰度高且表面可及。例如,IgG1抗体含有约85个赖氨酸残基,其中40多个通常可修饰,导致DAR的变异性更大 [22] 。综上所述,腙键连接子的稳定性不足和50%Gemtuzumab ozogamicin的高DAR导致载荷在血浆中的过早释放,导致高毒性,从而在2010年被迫撤市。
Sacituzumab govitecan使用pH敏感的水解性连接子在半胱氨酸残基上偶联到抗Trop2抗体,平均DAR为7-8。半胱氨酸偶联克服了赖氨酸偶联的异质性问题,每个抗体固定有8个自由的半胱氨酸残基 [23] 。为了抵消高DAR的高疏水性导致的加速清除,Sacituzumab govitecan在连接子上加入了短的聚乙二醇基团,以增加亲水性,增强溶解度并最小化聚集和清除。
最佳效力的DAR取决于连接子载荷如何平衡药物负载和体内行为。为此,不断开发新策略以增强ADC的效力,同时最大限度地提高ADC的稳定性。
总 结
ADC的复杂结构结合了单克隆抗体和小分子药物的特性,使临床医生在评估这些药物的安全性和有效性时面临独特的挑战。
由于这些药物使用了原本难以耐受的强效细胞毒药物,了解各种分子种类的药代动力学和药效学特性(包括ADC偶联物、总抗体和未偶联载荷)在靶组织和全身循环中的表现非常重要。尽管大多数剂量限制性毒性与载荷有关,但抗体和连接子也必须考虑在内,因为非特异性药物摄取、药物负载的异质性和载荷的过早释放等因素都可能影响毒性。
下期预告:
ADC药物常见不良反应——血液毒性
其他ADC内容专栏 :
1
参考文献(上下滑动查看):
* 此文仅用于向医学人士提供科学信息,不代表本平台观点
链接:http://www.lewenyixue.com/2024/10/18/ADC%E8%8D%AF%E7%89%A9%E5%AE%89%E5%85%A8%E6%80%A7%E7%AE%A1%E7%90%86%E2%80%94%E2%80%94%E7%9B%B8%E5%85%B3%E6%AF%92%E6%80%A7%E6%A6%82%E8%BF%B0/