



AI大模型助力智能化药物递送研发

在本研究中,论文团队通过自研大语言模型UVGPT和含时密度泛函理论TDDFT量子化学计算验证,成功设计出更有效的紫外光响应给药分子。 本研究为药物递送领域提出了新的大模型赋能的计算化学解决方案。
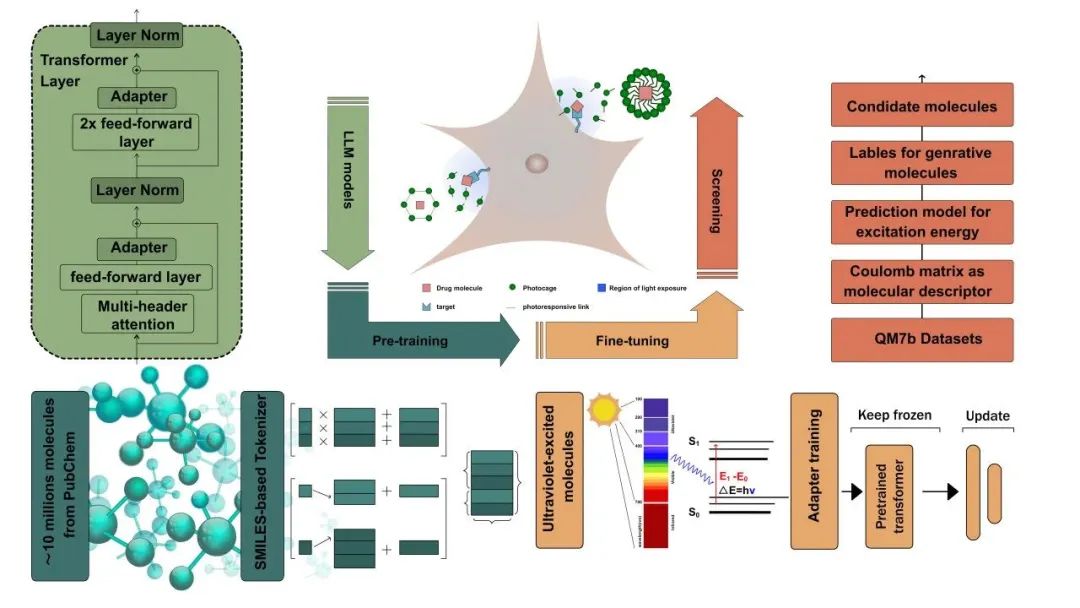
以此为基础使用紫外光分子数据集进行微调,使模型具备生成具有紫外光吸收特性的分子的能力。基于生成的紫外光分子数据集,研究团队进一步构建了筛选模型。团队使用了开源QM7b数据集用于微调UVGPT和训练筛选模型的分子激发能以及分子的库仑矩阵。UVGPT利用了包括激发能从4.13到12.41 eV之间的分子的训练数据集,并从中学习紫外光响应分子的定量结构-活性关系。
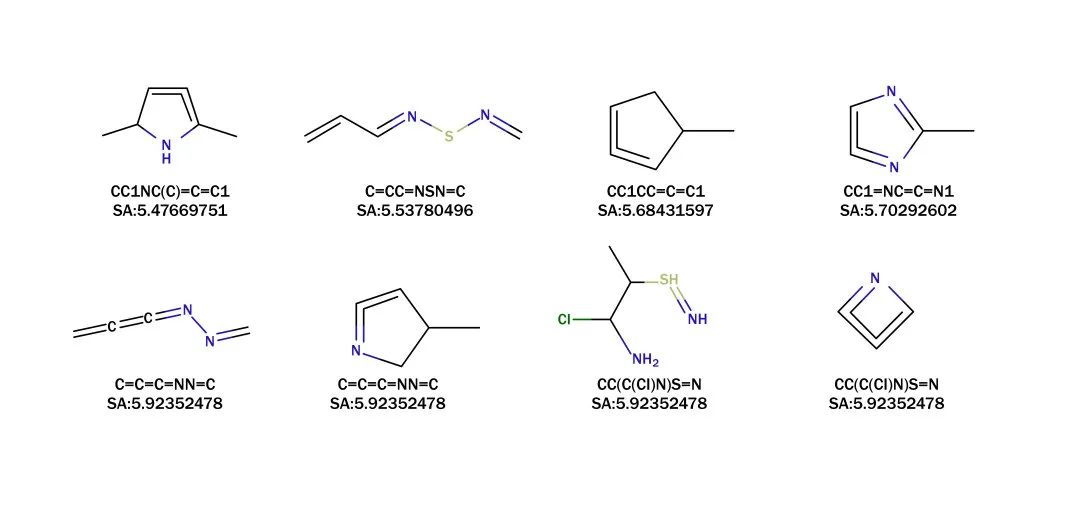
研究利用库仑矩阵作为分子描述符,预测分子的激发波长,并通过量子化学模拟来验证激发态特性。类药性评估以类药性的定量估计(QED)为指标,得分越高表明成药的可能性越大。合成可及性(SA)评分则用于系统性评估合成类药分子的难易程度,从而帮助确定分子设计化合物的优先次序。
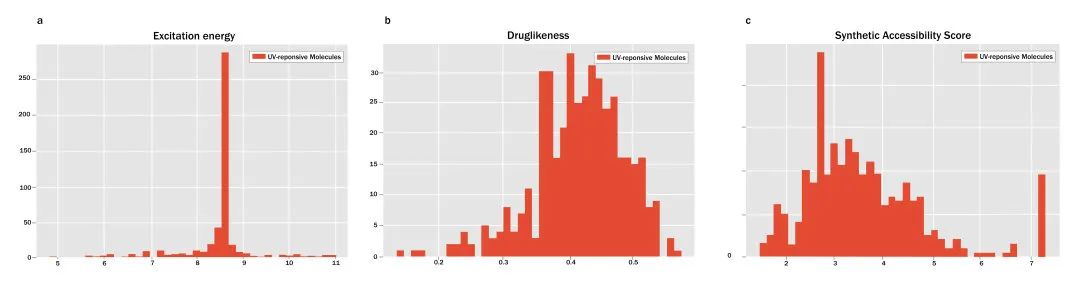
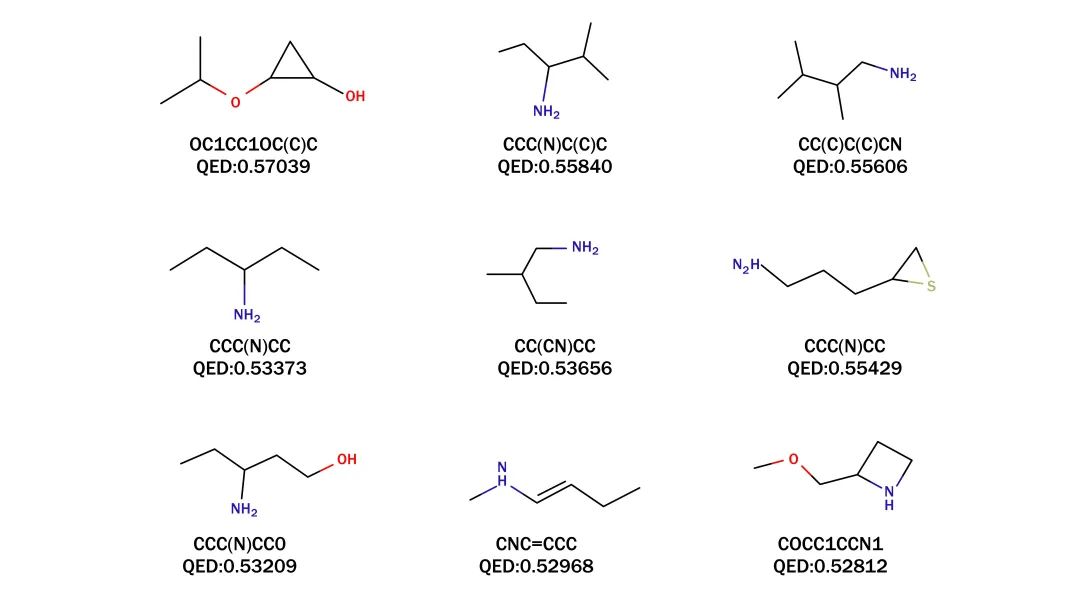
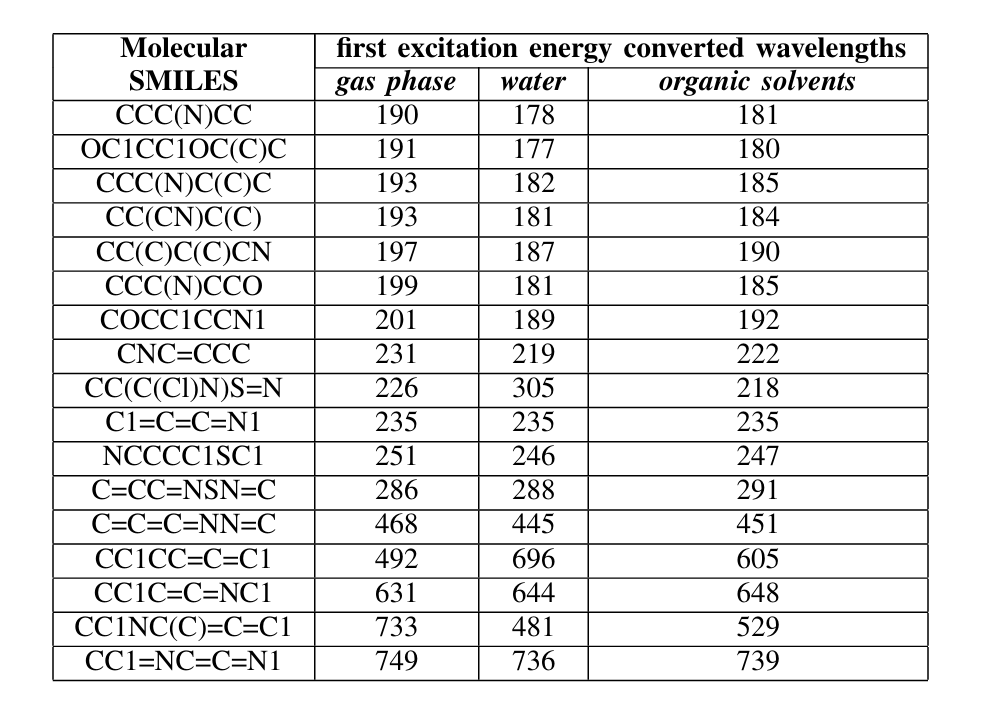
为了完善对分子激发能的评估,研究团队进行了量子化学计算(含时密度泛函理论TDDFT)。图2b显示了分子的QED排名。研究团队据此确定了9个类药性最高的分子,并在图3中展示了它们相应的SMILES分子式和QED值(从0.528到0.57不等)。值得注意的是,SMILES表达式为OC1CC1OC(C)C分子展现了最高程度的类药性,但它并不存在于PubChem数据库中,而PubChem中有一种化合物((1S,2S)-2-methylcyclopropan-1-ol)与OC1CC1OC(C)C相似,这进一步证明了该框架的有效性。
https://chemrxiv.org/engage/chemrxiv/article-details/65c2b0ee66c13817291af538
版权声明:本文为“乐问号”作者或机构在乐问医学上传并发布,仅代表该作者或机构观点,不代表乐问医学的观点或立场,不能作为个体诊疗依据,如有不适,请结合自身情况寻求医生的针对性治疗。
链接:http://www.lewenyixue.com/2024/03/22/ai%e5%a4%a7%e6%a8%a1%e5%9e%8b%e5%8a%a9%e5%8a%9b%e6%99%ba%e8%83%bd%e5%8c%96%e8%8d%af%e7%89%a9%e9%80%92%e9%80%81%e7%a0%94%e5%8f%91/
链接:http://www.lewenyixue.com/2024/03/22/ai%e5%a4%a7%e6%a8%a1%e5%9e%8b%e5%8a%a9%e5%8a%9b%e6%99%ba%e8%83%bd%e5%8c%96%e8%8d%af%e7%89%a9%e9%80%92%e9%80%81%e7%a0%94%e5%8f%91/
THE END
0
分享
海报
发表评论

赶快来坐沙发